





Volume 190 - Issue 11
Molecular biomarkers to individualise treatment: assessing the evidence
Authors: Chee K Lee, Sarah J Lord, Alan S Coates and R John Simes
Med J Aust 2009; 190 (11): 631-636. || doi: 10.5694/j.1326-5377.2009.tb02592.x
Published online: 1 June 2009
Published online: 1 June 2009
Abstract
The absolute benefit of a treatment varies between individuals depending on their prognosis before treatment and whether their response to the treatment varies from the overall relative risk reduction measured in clinical trials.
Based on these principles, biomarkers that can provide information about an individual’s prognosis or predict his or her treatment response can be used to tailor treatment decisions to individual patients.
Many novel molecular biomarkers are currently available. Although there is evidence to show that some of these can improve patient outcomes through improved biomarker-guided treatment strategies, others are yet to be adequately evaluated.
Randomised controlled trials (RCTs) can distinguish whether a biomarker provides prognostic or predictive information and assess whether using a biomarker to guide treatment improves patient outcomes.
Targeted RCTs can be used to demonstrate the efficacy of treatment in a restricted biomarker-defined population, and non-targeted RCTs can compare biomarker-guided versus conventional test-guided treatment strategies in broader populations.
The development of rapid high-throughput technologies to analyse DNA, RNA and protein has led to a paradigm shift in our understanding of the molecular basis of disease. Molecules that can be used to differentiate between normal and abnormal biological processes or predict treatment responses are termed “molecular biomarkers” (Box).1 Examples include DNA sequence mutations, epigenetic changes, and levels of messenger RNA or protein expression that are associated with a patient’s risk of disease events, survival time or response to treatment. Use of molecular biomarkers to supplement or replace conventional clinicopathological factors has the potential to transform the practice of medicine by creating new opportunities for developing and tailoring treatments to individual patients.
The clinical value of some molecular biomarkers has been established by trials that have demonstrated that biomarker-guided treatment strategies improve patient outcomes. For example, randomised controlled trials (RCTs) have provided evidence about the value of measuring oestrogen receptor (ER) status in women with breast cancer to identify those who will benefit from targeted treatment with anti-oestrogen therapy and those who will not. However, many molecular biomarkers discovered in observational studies are yet to be adequately evaluated in clinical practice.
In this article, we describe the principles of using biomarkers to individualise treatment and discuss the role of RCTs in assessing their clinical value.
What is individualised treatment?
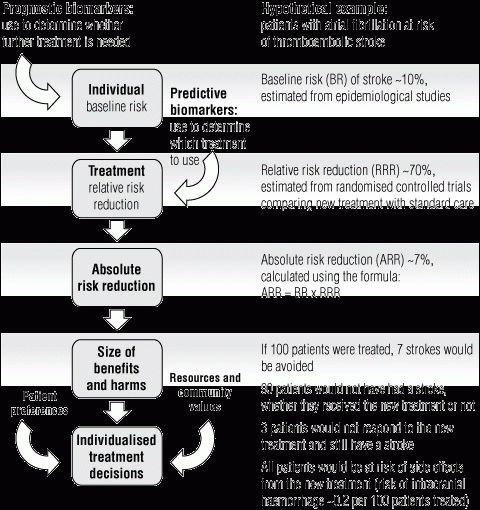
We rely on RCTs to identify the most effective treatment for patients with a given condition. Classically, these trials randomly assign patients to new or standard treatment and compare the risk of disease events, or time to disease events, to measure the effectiveness of the new treatment as a relative risk or hazard ratio. The absolute risk reduction for individual patients depends on their baseline risk before treatment — that is, their prognosis — and whether their response to treatment varies from the overall effect measured in RCTs — the “treatment prediction” (Box 2). If we have access to this information, we can individualise treatment decisions by weighing up the size of benefits and harms of the new treatment for individual patients.3
The full potential of using biomarkers to individualise treatment is not yet well understood. For example, trials have shown that anticoagulant therapy for the prevention of thromboembolic stroke in patients with non-valvular atrial fibrillation is more effective than standard care, but this does not mean that all patients will benefit. On the basis of trial results showing that anticoagulant therapy leads to a 70% relative risk reduction for major stroke, we can estimate that if 100 patients with a baseline risk of stroke of 10% were treated, seven strokes would be avoided.3 However, some patients would not have had a stroke, regardless of whether they received the anticoagulant therapy, and others would not respond to the anticoagulant therapy and still have a stroke (90 and three patients, respectively), but both groups might still experience the side effects of treatment. Ideally, a biomarker or panel of biomarkers could be used to identify the small subgroup of patients most likely to receive a clinical benefit — those who would otherwise most certainly have a stroke (the perfect prognostic test) and also respond to treatment (the perfect predictive test) — and offer treatment only to this group. This would obviate adverse events and unnecessary costs of treatment for low-risk patients and provide the opportunity to offer an alternative therapy to high-risk patients who would not be responsive to this treatment.
Identifying biomarkers that classify prognosis or
predict response
When a biomarker is discovered to be a promising classifier of patient outcome, further studies are needed to validate its reliability in independent patient populations and its discriminatory power compared with conventional clinicopathological tests.4-6 Its clinical value can then be determined by considering how this information will be used to guide treatment. This will depend on whether it can be used to classify patient prognosis, predict treatment response, or both. This information is not usually available from initial biomarker discovery studies, which are often undertaken retrospectively using specimens collected from a convenience sample of patients who have received no treatment, the same treatment, or a wide range of treatments.
Single-arm studies conducted in untreated populations can be used to identify prognostic biomarkers; however, RCTs are needed to determine whether these biomarkers can also be used to predict treatment response and how they may best be used to guide treatment. This is illustrated by the use of conventional biomarkers to guide decisions about cholesterol-lowering therapy. Large epidemiological studies have demonstrated the prognostic value of serum cholesterol to predict a patient’s risk of cardiovascular events,7,8 and RCTs have demonstrated that cholesterol-lowering therapy can reduce these risks.9 This leads to the question of whether serum cholesterol level or other conventional prognostic biomarkers can also predict who will respond to this treatment. To answer this question, RCTs that are large enough to compare treatment effects between biomarker-defined patient subgroups are needed. These comparisons use a simple statistical test for interaction. Alternatively, more sophisticated statistical methods can be used to explore the relationship between biomarker test results as a continuous variable and treatment effects.10,11
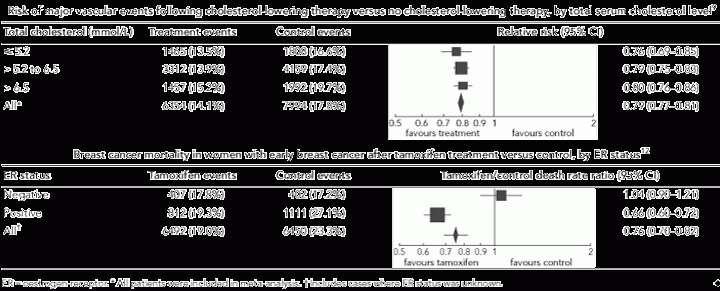
The Cholesterol Treatment Trialists’ prospective meta-analysis of cholesterol-lowering trials confirmed the prognostic value of conventional biomarkers (total serum cholesterol, age, presence of hypertension, etc) but found that none of these biomarkers provided additional information to predict treatment response (ie, no biomarker–treatment interaction).9 For example, patients with the highest cholesterol level (> 6.5 mmol/L) had the highest risk of major vascular events, but all patients, including those at the lowest risk (cholesterol level, < 5.2 mmol/L), benefited from cholesterol-lowering treatment; a relative risk reduction for major vascular events of around 20% in each risk group was reported (Box 3). Given similar treatment effects across different risk groups, patients at the highest risk will gain the greatest absolute benefit from treatment in terms of the total number of events avoided. Thus, prognostic information can be used to target treatment to patients at sufficiently high risk of events that the size of the absolute benefits justifies the costs and potential harms of the treatment.
Some biomarkers can provide both prognostic and predictive information, and this is often the case in cancer. An example is the ER status in breast cancer. Trial data from patients assigned to surgery without systemic adjuvant therapy have indicated that ER-positive tumours are associated with better early survival than ER-negative tumours; however, the difference in prognosis by ER status is relatively small and time-dependent.13,14 It was the discovery, through RCTs, that ER status predicts response to tamoxifen that transformed the use of anti-oestrogen therapy in the management of breast cancer. The Early Breast Cancer Trialists’ Collaborative Group’s meta-analysis of tamoxifen trials showed that tamoxifen reduced the risk of death among women with ER-positive tumours but not among women with ER-negative tumours (test for interaction, P < 0.00001; Box 3).12
Trial design for biomarker evaluation
When the potential role of a biomarker to guide treatment has been defined, its clinical value can be assessed by following the same rules of evidence that apply to the assessment of new treatments. Ideally, an RCT would be undertaken to test the hypothesis that the proposed biomarker-guided treatment strategy improves patient outcomes compared with the conventional testing strategy. Sometimes this information is available from existing RCTs of treatment. For example, tamoxifen trials have provided evidence about the difference in patient outcomes if tamoxifen is reserved for patients with ER-positive breast tumours compared with the scenario without ER testing where treatment may be given to all women (or none).12 One exception where an RCT may not be justified is when a prognostic biomarker can accurately identify patients at low risk of disease who can safely avoid treatment. In this situation, single-arm studies demonstrating the prognostic accuracy of the biomarker versus conventional tests may suffice.
Classical trials
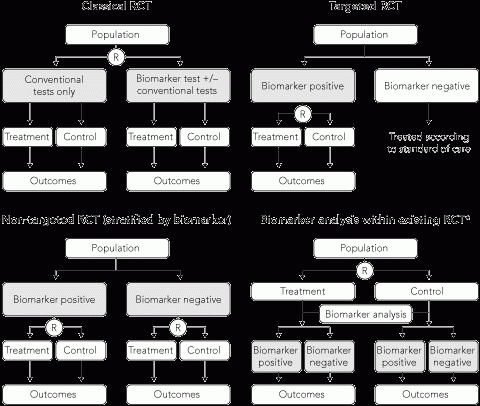
Using a classical RCT design, the impact of treatment with the availability of a novel biomarker can be tested by randomly assigning patients to either the biomarker test with or without conventional tests, or conventional tests only (Box 4). This design evaluates the effectiveness of the combination of the new biomarker test and the subsequent treatment intervention. However, it is an inefficient design as a large number of patients are needed but only patients with a biomarker test result that disagrees with the conventional test result contribute to the expected outcome from the treatment intervention.15 All patients testing positive by both investigations will receive the same treatment intervention, hence the biomarker does not discriminate outcomes for these patients.
Targeted trials
An alternative approach is a targeted RCT,16,17 also referred to as an enrichment design, which involves upfront testing of all patients for the biomarker of interest and selecting only patients with biomarker results that will lead to a change in outcome with the proposed treatment. This is more efficient than a classical design as those without the biomarker of interest are excluded from the study, hence fewer patients are enrolled (Box 4). A positive trial result provides evidence about the effectiveness of using the biomarker to guide treatment. The design is generally used when there is robust a priori biological evidence that treatment is effective or more effective in the biomarker-defined population.
As targeted trials recruit patients with the same biomarker status, they can be used to assess the efficacy of a new treatment in a biomarker-defined population, or the efficacy of an existing treatment in a subgroup of patients newly defined by a biomarker. Recent oncology trials provide examples of these scenarios.
Testing a new treatment in a biomarker-defined population. The pivotal trial for trastuzumab, a monoclonal antibody against the human epidermal growth factor receptor 2 (HER2) protein, was undertaken in a targeted population of women with metastatic breast cancer who were positive for HER2 (Box 5). This trial showed the efficacy of the combined strategy of testing for HER2 status and treating women with HER2-positive tumours with trastuzumab.18 Subsequent research has focused on improving assay methods and the cut-off level of HER2 amplification or expression to optimise the predictive performance of HER2, to identify women who will benefit from treatment with trastuzumab.23
Testing an existing treatment in a subgroup of patients newly defined by a biomarker. Oncotype DX is a 21-gene prognostic assay developed to classify women with node-negative, ER-positive breast cancer into three categories according to their risk of developing recurrent disease (low, intermediate and high risk). It has been proposed to guide treatment decisions by sparing women who are at low risk unnecessary chemotherapy, and identifying those who are at high risk and need treatment. Oncotype DX is currently being prospectively assessed in the TAILORx trial (Trial Assigning Individualized Options for Treatment [Rx]).19 The primary objective of the trial is to investigate the efficacy of chemotherapy as an addition to hormone therapy in women who are at intermediate risk (recurrence score, 11–25) (Box 5). The working premise is that patients in the intermediate-risk group will do no worse with hormone therapy alone than they would with hormone therapy plus chemotherapy. This study assumes that chemotherapy does not improve outcomes in patients at low risk (recurrence score, < 11) but will be beneficial in patients at high risk (recurrence score, > 25).
Targeted RCTs offer an efficient method to show proof of concept for the efficacy of treatment in biomarker-selected patient groups. They can be used where there is a biological assumption that only a biomarker-defined patient group will benefit from the new treatment; hence restricting the trial to this group (eg, biomarker-positive patients) is more efficient and potentially more ethical. However, molecular pathways of tumour pathogenesis are complex and often not clearly understood, so questions about the efficacy of treatment in biomarker-negative patients may also warrant consideration. For example, trastuzumab was recently reported to have efficacy in HER2-negative patients in a retrospective analysis of a broader patient population that included both HER2-positive and HER2-negative patients.24
Non-targeted trials
In contrast to targeted trials, non-targeted (or unselected design) trials do not restrict recruitment to a single biomarker-defined subgroup of patients. This design is needed to assess the predictive value of a biomarker to distinguish between patients who will respond differently to the same treatment (Box 4), or compare the efficacy of a new biomarker-guided treatment strategy with current best practice using conventional testing.
The International Breast Cancer Study Group Trial IX, which randomly assigned postmenopausal node-negative early-stage breast cancer patients, stratified according to ER status, to chemotherapy followed by tamoxifen versus tamoxifen alone, is an example of the first situation. An example of the second situation is the ongoing MINDACT (Microarray In Node-negative Disease may Avoid ChemoTherapy) trial, which is comparing the prognostic value of a 70-gene signature for breast cancer with established clinicopathological criteria to identify women with node-negative early-stage breast cancer who can safely avoid adjuvant chemotherapy (Box 5). Unlike the TAILORx trial, the MINDACT trial does not restrict randomisation to one biomarker-defined patient subgroup. All women are assessed by using both the new biomarker gene signature and conventional criteria to classify their risk of disease recurrence. Only women with discordant results (biomarker-negative and conventional criteria-positive, or vice versa) are randomly assigned to receive chemotherapy or no chemotherapy; women with concordant results are not randomised but treated according to the standard of care (women at low risk with observation, and women at high risk with chemotherapy). This trial will provide data that compare patient outcomes when chemotherapy is selected according to the biomarker versus conventional criteria.
Non-targeted trials generally require a large sample size, particularly if the incremental effects of offering treatment to patients reclassified using a biomarker are small. Hence, their main disadvantage is that they may not be feasible if a small proportion of patients tested with the target condition are eligible — for example, if biomarker prevalence is low. Design variations to optimise the efficiency of non-targeted trial designs are discussed elsewhere and are beyond the scope of this article.17
Biomarker analysis within an
existing RCT
A pre-specified analysis of prospective data collected from an existing RCT of treatment can sometimes be used to test a hypothesis about the prognostic or predictive value of a biomarker (Box 4). This study design was used to assess K-ras status as a predictor of response to panitumumab, an epidermal growth factor receptor inhibitor, in patients with metastatic, chemotherapy-refractory colorectal cancer. Investigators used archival tissue from an earlier RCT, which demonstrated the efficacy of panitumumab compared with best supportive care in a non-targeted population, to test the hypothesis that K-ras status predicts treatment response (Box 5).22,25
This study design has the advantage of time and cost efficiency. Its major disadvantage is the potential for selection bias if archival tissue or serum data are not available for all patients. Other disadvantages include measurement bias if measurements of the biomarker or statistical analyses are not blinded, and chance false-positive findings due to multiple comparisons, particularly with trial data used to explore multiple candidate biomarkers. These problems reduce the validity of study results, so analyses should only be considered if the biomarker can be measured on all or a large representative sample of all patients. Furthermore, the design needs a biologically plausible hypothesis, a prospectively defined protocol for the biomarker assays, and statistical analysis plans.
Implications for future trials
The current availability of genomic, transcriptomic, proteomic, metabolic and other similar technologies provides unprecedented opportunities for individualised treatment through the discovery of molecular biomarkers and the development of molecularly targeted therapies. The conventional rules of evidence for evaluating these new technologies have not changed, but the need for more efficient RCTs has led to innovations in the design of targeted and non-targeted trials. If an existing treatment trial has archived specimens from all, or most, participants, then an efficient and reliable evaluation of a new biomarker may be achievable by further analysis of these specimens. Future trials should be designed to anticipate these data needs.
1 Definitions
Biomarker: a “characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacological responses to a therapeutic intervention”.1 Molecular biomarkers include genetic factors measured as variations or mutations of DNA sequence, epigenetic factors, and levels of messenger RNA and proteins.
Prognostic biomarker: classifies an individual’s baseline risk of having a clinical event.
Predictive biomarker: classifies the magnitude of an individual’s response to treatment.
2 Role of biomarkers to classify patient prognosis or predict treatment response for individualised treatment decisions*
 | |||||||||||||||
|
* Flow chart reproduced with permission from Lord S, Lee C, Simes RJ. The role of prognostic and predictive markers in cancer. Cancer Forum 2008; 32 (3): 139-142;2 hypothetical example based on clinical scenario from Glasziou PP, Irwig LM. An evidence based approach to individualising treatment. BMJ 1995; 311: 1356-1359.3 | |||||||||||||||
4 Randomised controlled trial (RCT) designs for biomarker evaluation
 | |||||||||||||||
|
R = randomisation. * Research hypotheses may be pre-specified in the trial protocol (prior to study unblinding) or defined retrospectively. | |||||||||||||||
5 Purposes of randomised controlled trials (RCTs) used to evaluate biomarkers: recent examples in oncology
Competing interests
None identified.
Acknowledgements
We thank Rhana Pike for assistance in preparation of this article.
References
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 2001; 69: 89-95. 0_CHDIJJBC
- Lord S, Lee C, Simes RJ. The role of prognostic and predictive markers in cancer. Cancer Forum 2008; 32 (3): 139-142. 0_CHDJHBJD
- Glasziou PP, Irwig LM. An evidence based approach to individualising treatment. BMJ 1995; 311: 1356-1359. 0_i1092191
- Hayes DF, Bast RC, Desch CE, et al. Tumor marker utility grading system: a framework to evaluate clinical utility of tumor markers. J Natl Cancer Inst 1996; 88: 1456-1466. 0_i1092193
- Pepe MS. Evaluating technologies for classification and prediction in medicine. Stat Med 2005; 24: 3687-3696. 0_pgfId-1098900
- Ransohof DF. How to improve reliability and efficiency of research about molecular markers: roles of phases, guidelines, and study design. J Clin Epidemiol 2007; 60: 1205-1219. 0_i1092196
- Rose G, Shipley M. Plasma cholesterol concentration and death from coronary heart disease: 10 year results of the Whitehall study. Br Med J (Clin Res Ed) 1986; 293: 306-307. 0_i1092198
- The Pooling Project Research Group. Relationship of blood pressure, serum cholesterol, smoking habit, relative weight and ECG abnormalities to incidence of major coronary events: final report of the pooling project. J Chronic Dis 1978; 31: 201-306. 0_i1092200
- Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366: 1267-1278. 0_i1092202
- Bonetti M, Gelber R. A graphical method to assess treatment–covariate interactions using the Cox model on subsets of the data. Stat Med 2000; 19: 2595-2609. 0_i1092204
- Royston P, Sauerbrei W. A new approach to modeling interactions between treatment and continuous covariates in clinical trials by using fractional polynomials. Stat Med 2004; 23: 2509-2525. 0_i1092206
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005; 365: 1687-1717. 0_i1092208
- Saphner T, Tormey DC, Gray R. Annual hazard rates of recurrence for breast cancer after primary therapy. J Clin Oncol 1996; 14: 2738-2746. 0_i1092210
- Fisher B, Redmond C, Fisher ER, Caplan R. Relative worth of estrogen or progesterone receptor and pathologic characteristics of differentiation as indicators of prognosis in node negative breast cancer patients: findings from National Surgical Adjuvant Breast and Bowel Project Protocol B-06. J Clin Oncol 1988; 6: 1076-1087. 0_i1092212
- Bossuyt PM, Lijmer JG, Mol BW. Randomised comparisons of medical tests: sometimes invalid, not always efficient. Lancet 2000; 356: 1844-1847. 0_i1092214
- Sargent DJ, Conley BA, Allegra C, Collette L. Clinical trial designs for predictive marker validation in cancer treatment trials. J Clin Oncol 2005; 23: 2020-2027. 0_i1092216
- Simon R, Maitournam A. Evaluating the efficiency of targeted designs for randomized clinical trials. Clin Cancer Res 2004; 10: 6759-6763. 0_i1092218
- Salmon DJ, Leyland-Jone B, Shak S. Use of chemotherapy plus a monoclonal antibody against HER2 positive metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344: 783-792. 0_i1092220
- Sparano JA, Paik S. Development of the 21-gene assay and its application in clinical practice and clinical trials. J Clin Oncol 2008; 26: 721-728. 0_i1092222
- International Breast Cancer Study Group (IBCSG). Endocrine responsiveness and tailoring adjuvant therapy for postmenopausal lymph node-negative breast cancer: a randomized trial. J Natl Cancer Inst 2002; 94: 1054-1065. 0_i1092224
- Bogaerts J, Cardoso F, Buyse M, et al. Gene signature evaluation as a prognostic tool: challenges in the design of the MINDACT trial. Nat Clin Pract Oncol 2006; 3: 540-551. 0_i1092228
- Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359: 1757-1765. 0_i1092229
- Wolff AC, Hammond ME, Schwartz JN, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol 2007; 25: 118-145. 0_i1092231
- Paik S, Kim C, Wolmark N. HER2 status and benefit from adjuvant trastuzumab in breast cancer. N Engl J Med 2008; 358: 1409-1411. 0_i1092233
- Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26: 1626-1634. 0_i1092235