





Volume 186 - Issue 3
Australia’s century of meningococcal disease: development and the changing ecology of an accidental pathogen
Author: Mahomed S Patel
Med J Aust 2007; 186 (3): 136-141. || doi: 10.5694/j.1326-5377.2007.tb00837.x
Published online: 5 February 2007
Published online: 5 February 2007
Abstract
Trends in meningococcal disease (MD) over the 20th century in Australia, as in other industrialised countries, have been characterised by epidemics during the two World Wars, a transient rise in incidence in the 1950s followed by endemic disease, and in the 1980s the emergence of a sustained hypersporadic phase. Epidemics occur at times of social upheaval and among marginalised populations, and resolve when living conditions improve.
Periodic serogroup A epidemics have been replaced since the 1950s by endemic disease caused mainly by serogroups B and C meningococci. The current hypersporadic plateau in Australia, as in other industrialised countries, is associated with the intercontinental spread of hypervirulent clones of meningococci.
The conjugate serogroup C vaccine has reduced the incidence of MD and carriage rates of serogroup C meningococci. However, the vaccine is expensive and its long-term impact on the emergence of non-vaccine strains and on nasopharyngeal microecology is unknown.
A rising incidence of MD should not be viewed as the action of a virulent microbe exploiting a vulnerable population, but as the emergence of an “accidental pathogen” from an evolving host–microbial ecology. While it is essential to monitor the impact of vaccines on this ecology, we must find ways that can optimise our coexistence with microbes.
In 2003, Australia introduced routine childhood and adolescent vaccinations against meningococcal disease (MD) caused by serogroup C (SGC) meningococci. In its first year, the program cost an estimated $41 million, and the government is committed to continue funding it for the next 14 years.1 National vaccination programs to control the rising incidence and mortality from SGC-MD with the new conjugate vaccine were pioneered in the United Kingdom in 1999, and have also been adopted in the Netherlands, Spain, Belgium, and Canada.2,3
While the vaccine has effectively reduced the incidence of SGC-MD, it has also reduced the carriage of SGC meningococci.3 A similar suppressive effect on disease incidence and carriage associated with the respective vaccine strains was observed after the introduction of the conjugate Haemophilus influenzae type b and pneumococcal vaccines.4,5 To anticipate the kinds of changes we might see with mass vaccinations against SGC-MD, we need to understand the changing epidemiology of MD. (Terms are defined in the Glossary.)
Neisseria meningitidis has no host other than humans, so the history of its changing ecology mirrors, in part, that of its human hosts. Yet there exists no social history of MD. Here, I draw on the epidemiological history of the disease in Australia over the past century to answer two questions: What were the evolving trends in human–microbial ecology that led to the new vaccination policy? How may the conjugate meningococcal vaccine impact on human–microbial ecology, and how should we monitor this impact?
What were the evolving trends in human–microbial ecology that led to the new vaccination policy?
Understanding the changing epidemiology and ecology of MD requires the linkage of incidence data with microbiological data. Long-term incidence data for MD are lacking in all countries. However, routine surveillance data can act as a proxy, if limitations of these data are understood. In Australia, annual notification data for MD have been collected since 1915.6-8 While limited microbiological data have been available since 1960,9-12 national data were not collected systematically until 1994.13-22 In addition, new molecular technologies can now be used to track the temporal spread of meningococcal strains across continents.23-26 Synthesis of these data enables us to make some statements about the changing microbial–human ecology.
Shift from epidemic to endemic to hypersporadic patterns of disease
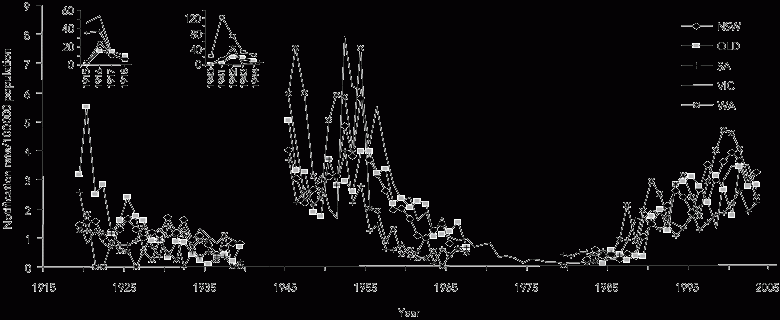
Annual notification rates of MD in Australia from 1915 to 2003 adapted from three data sources6,7,27 are summarised in Box 1 and Box 2. Although there was no explicit surveillance definition for MD, and the states had independent systems of collecting and reporting notifications before 1991, the trends are remarkably consistent.
Nationwide epidemics occurred during the two World Wars as part of the pandemics associated with military mobilisation and social disruption.30,31 The only other epidemics since then have been restricted almost entirely to Indigenous people in Central Australia.32,33
After World War II, notifications rose in the early 1950s in parallel with those in other industrialised countries.31,34,35 This period was associated with extensive population movements within Europe and migration to North America, Australia and New Zealand. Notifications then dropped consistently across all states to a nadir of less than 0.5/100 000 population by 1981, but rose again in 1987 to be sustained by a hypersporadic plateau that led ultimately in 2003 to the vaccination program against SGC-MD.1
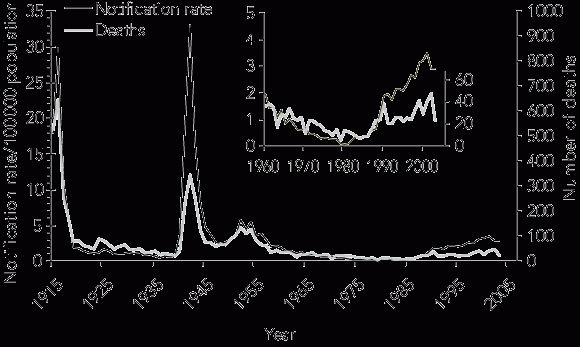
Is this change in disease pattern reflected in the pattern of deaths due to MD? Until 1990, the trend in the number of deaths from MD29 mirrored the trend in notifications (Box 2). Reasons for the divergence after 1990 (an observation also noted in the UK36) may include improved case ascertainment, earlier diagnosis and treatment, and reduced frequency of life-threatening disease.
Routine surveillance for MD, as for other notifiable diseases, underestimates disease incidence. However, the consistent secular trends in routine surveillance and mortality data, as well as data from laboratory-based9-11 and incidence studies33 (Dr Rob Condon, formerly Infectious Disease Epidemiologist, Telethon Institute for Child Health Research, Perth, WA, personal communication) in the latter part of the last century, provide compelling evidence of the change in epidemiology since the late 1980s. The trends resemble those of other industrialised countries.26,31,34,35,37
The fall of serogroup A meningococcal disease
Epidemic and hypersporadic MD are triggered typically by the introduction and spread of a single hypervirulent clone of meningococci,23-26 and curtailed naturally once herd immunity develops and the pool of people susceptible to the strain is exhausted.
The pandemics of the World Wars were caused by serogroup A meningococci (SGA). Disease incidence caused by this serogroup declined subsequently, to be replaced mainly by serogroup B meningococci (SGB) and SGC.23,24,26,31,35 This shift was almost complete in industrialised countries by the second half of the 20th century when it accounted for less than 1% of people with MD.9,10,26,31
There were two notable exceptions. The first, the 1971–1973 epidemic in Central Australia (encompassing adjacent regions of the Northern Territory, South Australia and Western Australia),32 was concurrent with epidemics caused by clonal subgroup I of SGA in Africa and the Middle East, and with sporadic disease in many other countries,23,25 including an outbreak among Native Americans.38 The second, the 1987–1991 epidemic in Central Australia,33 was also caused by this clonal subgroup;23 the same strain was responsible for the 1985–1987 epidemic in Auckland, NZ, with very high attack rates among Polynesian and Māori children.26,39 SGA-MD has not recurred in Central Australia since the epidemic ended in 1991.
The rise of serogroups B and C meningococcal disease
Although Australian microbiological data are not available for the 1950s, in the UK and the United States, a contemporaneous rise in incidence was associated with SGB- and SGC-MD, accounting for 60%–80% and 20%–33% of cases, respectively.31,35 Microbiological data available for endemic disease between 1960 and the 1980s in Australia9-12 indicate that SGC emerged as an important pathogen in the 1980s, together with the rise in SGB, the dominant pathogen of the 1960s and 1970s. The first cluster of cases of SGC-MD (five cases) was reported in 1968 from Canberra.10 In Victoria, data on admissions to Melbourne’s Royal Children’s Hospital since 1960 reveal that SGC-MD was first detected in 1982,9 and its incidence increased from a third of cases in 1985 to 15 of 25 cases in 1989, the remaining cases being associated mainly with SGB-MD. In South Australia, while SGC-MD has been recorded since data collection started in 1971, its incidence also increased sharply after 1984, and in 1989 exceeded the incidence of SGB-MD for the first time.10,11 Data collected in New South Wales between 1977 and 1987 show cases of SGC-MD for the first time in 1980.12 In Western Australia, data collected since 1984 show that, concurrent with the Central Australian SGA-MD epidemic, SGC-MD increased from three of 13 cases in 1986 to peak at 26 of 44 cases in 1989 (Dr Rob Condon, formerly Infectious Disease Epidemiologist, Telethon Institute for Child Health Research, Perth, WA, personal communication). The corresponding number of cases of SGB-MD were one and four, respectively, but this serogroup has accounted consistently for over 60% of cases since 1991.13-22
Thus, while SGB-MD predominated in the 1960s and 1970s, SGC started emerging as an important pathogen in the 1980s. The transition from the endemic to hypersporadic phases in the late 1980s was associated with an increase in disease caused by both serogroups. Between 1989 and 1995, cluster A4 (the clonal complex associated mainly with C:2b:P1.2 [see Glossary]) caused small multifocal outbreaks in Katanning (WA) and south-western Sydney, and in Aboriginal communities in North Queensland.40 Invasive isolates from these outbreaks and from sporadic cases were closely related to each other genetically,41 suggesting a common point of introduction into Australia. Of the 58 cases of MD in south-western Sydney between 1990 and 1994, SGC (mainly C:2b:P1.2,5) accounted for 31 of 58 invasive isolates, and SGB meningococci for 16 isolates.42
Through the 1990s, the incidence of MD due to the ET-37 complex (associated mainly with C:2a:P1.2,5) and its variants also increased. It was responsible for the outbreak connected with a Penrith (Sydney) nightclub in 1996,43 and the peak in incidence in Victoria in 1999.44
In contrast to the fluctuating incidence of SGC-MD, the incidence of SGB-MD due mainly to the ET-5 complex (associated with B:15:P1.6) and lineage III (associated with B:4:P1.4) persisted for over a decade, accounting for about two-thirds of all cases of MD in Australia.13-22 An effective vaccine against all strains of SGB is not available yet to control the incidence of SGB-MD.
Over the 5-year period before the conjugate SGC vaccine was introduced in 2003, the frequency of SGC-MD increased steadily in Victoria, Queensland and Tasmania, dropped consistently in New South Wales, and remained steady in South Australia, the Australian Capital Territory and the Northern Territory.17-21 The average annual incidence of SGC-MD over these 5 years was 1.1/100 000, but the extent to which the laboratory-based surveillance data reflect the overall incidence of SGC-MD is not known.
Concordant disease patterns across the industrialised world
Understanding changes in the local epidemiology and ecology of MD requires a global perspective. The post-1970 hypersporadic phase in other industrialised countries preceded and, in many ways, shaped the Australian experience. Hypersporadic disease was first reported in northern Norway in 1975, and was associated with the ET-5 complex.24 This clone spread across north-western Europe and later to the US.24 In the 1970s, cluster A4 was a common cause of MD in the US, Canada and the UK,24 and, in the 1990s, caused outbreaks in the UK and Spain.37 MD due to lineage III and the ET-37 complex also increased across Europe in the 1990s.24,37 In New Zealand, the incidence of MD increased tenfold in the 1990s due mainly to lineage III (B:4:P1.7b,4) associated with epidemic proportions in the Pacific and Māori populations.45
The hypersporadic plateau in industrialised countries therefore reflects the summation of a series of overlapping hypersporadic waves caused by the intercontinental spread of multiple hypervirulent clones of meningococci. While SGA-MD was highly restricted by time, place and population, the incidence of SGC-MD rose and fell rapidly over a few years, and the incidence of SGB-MD remained consistently high for well over a decade.
The evolving host–microbial ecology of MD in industrialised countries
Although disease incidence has been categorised as a triad of endemic, hypersporadic and epidemic MD, incidence occurs along a continuum. Temporal and spatial fluctuations along this continuum reflect variations in outcomes of the dynamically evolving host–microbial ecology. While host defences adapt to protect against tissue invasion, meningococci have amassed high levels of genetic variability to evade this defence repertoire46 and to colonise new hosts. They have evolved an ingenious “phase variation” potential (fine-tuning) to change surface structures that enable ongoing transmission and colonisation without having to invade tissues and cause disease.46 Invasion is not essential to survival; rather, it is an evolutionary dead-end for meningococci. Virulence emerges as an accidental by-product of the immune forces that select out genes coding for transmissibility.46 N. meningitidis, it therefore appears, has not evolved to cause disease but does so as an “accidental pathogen”.47
The replacement of SGA by SGB and SGC as the major pathogens of MD in industrialised countries reflects microbial co-adaptation as we modify the ways we live, behave and mix with each other across the globe. This changing ecology led to a shift away from periodic epidemics, except in vulnerable subgroups such as Australian Aboriginals and New Zealand Māori.32,33,39,45 Poverty, crowded households and neighbourhoods, and reduced access to health services represent socially constructed faultlines along which epidemics may erupt.26,32,33,36,38,48 While the demise of epidemic SGA-MD can be attributed to improved living conditions,26 the societal determinants of the current hypersporadic phase, in addition to factors related to poverty and crowding, are unclear. Understanding this requires an ecosystems approach to causation, one that looks upstream of molecular markers and individual disease risk factors.
Cost-effectiveness of the conjugate SGC vaccine
The report (including cost-effectiveness) and recommendations on the introduction of the conjugate SGC vaccine by the Australian Technical Advisory Group on Immunisation are not available in the public domain. However, cost-effectiveness considerations are believed to have been important in arriving at the recommendation for a single-dose program at age 12–13 months (current policy)1 rather than a three-dose program at birth (UK policy).3 A study in the Netherlands showed relatively high incremental costs per life-year saved for two- and three-dose programs at birth compared with a single dose at age 14 months,2 and supports the Australian recommendation.
How may the conjugate meningococcal vaccine impact on human–microbial ecology, and how should we monitor this impact?
The long-term impact of the conjugate meningococcal vaccine on host–microbial ecology is unclear. Conjugate vaccines against the encapsulated bacteria regulate the ecology of the nasopharynx.3-5 However, what remain unknown are the physiological and health benefits of the nasopharyngeal microflora and of the complex inter-relationships and interdependence that have evolved over centuries between the colonising microbes. This contrasts with our growing knowledge of the essential role of the gut microflora in the development and lifelong maintenance of generic immune functions and the architectural integrity of the gut.49
Evidence of effects of the meningococcal vaccine on human–microbial ecology
While the polysaccharide SGA and SGC vaccines were highly effective in controlling outbreaks of MD caused by these serogroups,50,51 they may have impacted adversely on disease caused by serogroups W135 (SGW135) and SGB. Epidemics caused by SGW135 have started to emerge in some African countries and, since 2000, carriage rates increased for the first time since this strain was initially detected in 1968.52 These events could, in part, be attributed to mass vaccinations against SGA-MD,52 with SGW135 emerging as a “replacement” strain. Disease caused by this serogroup also appears to have gained a foothold in Europe since 2000.52 After mass vaccinations with the polysaccharide SGC vaccine to control outbreaks in Spain and the Czech Republic, capsule switching occurred to shift the cause of MD from the C ET-37 complex to the B ET-37 complex.3,52 However, because similar capsule switching was also observed when the vaccine was not used,3 a direct cause–effect relationship to implicate the vaccine has not been confirmed.
Evidence of effects of other conjugate vaccines on human–microbial ecology
The conjugate H. influenzae type b vaccine reduced both disease and carriage rates. Initial concerns of an immediate replacement of type b by other strains have not been borne out; only a minimal increase in disease caused by the type a strain has been observed.4 The experience after the introduction of conjugate pneumococcal vaccine is different. While meningitis and bacteraemic pneumonia caused by the seven vaccine serotypes have nearly been eliminated, and carriage rates of vaccine strains have dropped,5 overall carriage rates of pneumococci have not changed much because of increased carriage of non-vaccine serotypes.5 Possible explanations for persistent carriage rates include unmasking of previous minority strains, replacement by serotypes unaffected by the vaccine, or a serotype switch to evade immunity.5 The prognosis for a lasting suppression of pneumococcal disease is guarded.5
Monitoring the impact of the conjugate SGC vaccine
Monitoring the changing incidence and epidemiology of MD should be linked with other initiatives, particularly clinical and population-based studies to monitor changing human–meningococcal ecology. From the disease perspective, we need to monitor not only changes in the age distribution, severity, and microbiology of MD across subpopulations, but also changes in respiratory tract infections with other microbes, such as the non-vaccine strains of H. influenzae4 and pneumococci,5,53 as well as the atypical infections such as Mycoplasma pneumoniae and Chlamydia pneumoniae.54 From the population perspective, we should monitor changes in the carriage of meningococci, as well as in the carriage of other nasopharyngeal microflora. In addition, we should improve our understanding of how microbes help program our immune systems from birth and, more specifically, our understanding of the type and timing of infections that promote the development of natural immunity against infections,49 including MD.55
Conclusion
Trends in MD over the 20th century fluctuated along an incidence continuum, with epidemics erupting at times of social upheaval and among marginalised populations.26,30,32-34,36,38,48 Epidemics gave way to endemic disease as living conditions improved,26 and although the societal determinants of the current hypersporadic pattern are unknown, this phase has been sustained by the spread of hypervirulent clones associated with increasing volumes of human (and microbial) traffic across the globe.
SGC emerged as an important pathogen in Australia in the 1980s. Over the 5 years before the conjugate vaccine was introduced, the annual incidence of SGC-MD fluctuated, and was particularly low in some states and territories, averaging 1.1/100 000 population nationally (based on laboratory-surveillance data). However, the vaccine is expensive, and its long-term impact on nasopharyngeal microecology and on the emergence of non-vaccine serogroups is unknown.
We should not view a rising incidence of MD as the action of a virulent microbe exploiting vulnerable human defences, but rather as a variant outcome of an evolving host–microbial ecology. Because the conjugate vaccines aimed at protecting us against diseases also regulate the microecology of the nasopharynx, their use must be regarded as an experiment in restructuring the local bacterial population biology.56 While we must monitor their impact on the evolving host–microbial ecology, we must also find ways to optimise our coexistence with microbes.49
Glossary
Endemic: The constant presence of disease; for meningococcal disease (MD) in industrialised countries, this is usually an annual incidence of < 2/100 000 population.
Hypersporadic: Incidence levels exceeding endemic but not as high as epidemic levels, with the occurrence of multifocal clusters of cases (small spatio-temporal group of cases distributed in locations around the country). In industrialised countries, an annual incidence of 2–10/100 000 population is considered hypersporadic; it has been associated with a high case-fatality rate (over 10%), particularly among teenagers and young adults. The term “hyperendemic”, used by some authors, is not used here as it suggests a disease that is constantly present at a “high” incidence.
Incidence continuum: The annual incidence of MD fluctuates along a range of values (continuum) from < 1/100 000 population to, say, 10 or 1000/100 000 population. The threshold values for distinguishing endemic from hypersporadic, and hypersporadic from epidemic levels, are relatively arbitrary.
Hypervirulent clone: Studies of the population genetics and molecular markers of thousands of meningococcal isolates from around the globe revealed how hypersporadic and epidemic levels were associated consistently with selected clones (lineages) of Neisseria meningitidis. These clones have been labelled “hypervirulent”.
Capsule switching: Meningococcal serogrouping (eg, A, B, C, Y, W135) is based on the properties of the outer capsule, a phenotypic expression of the capsule-specific gene. Horizontal transfer of this gene between different serogroups may result in the conversion (switch) in serogroup without changes in other genetic markers. This is an ingenious strategy used by meningococci and other encapsulated bacteria to evade natural or vaccine-induced immunity directed at the capsule.
Characterisation of meningococci: Serogrouping and serotyping are based on the properties of the outer capsule and outer membrane proteins (OMPs), respectively. Antigens of classes 2 and 3 OMPs appear as the second number (serotype) in the formula (eg, C:2a). Serosubtyping is based on the diversity of the class 1 OMP, and numbers are quoted after the prefix P1 (eg, C:2a:P1.2). These phenotyping methods are being replaced by molecular typing of the genome, and correlate closely with epidemiological patterns of MD, including geographical spread of specific clones. Multilocus enzyme electrophoresis (MLEE) was used to classify meningococci into clonal “subgroups” for serogroup A, into “complexes” or “clusters” for serogroups B and C, and also into clones for serogroup B. MLEE has been replaced by the more portable multilocus sequence typing (MLST) that characterises meningococci into clonal groups comparable with MLEE.
1 Annual notification rates of meningococcal disease in five states in Australia, 1915–2003
Data for Tasmania, the Northern Territory and the Australian Capital Territory are not shown because of the widely fluctuating incidence associated with their smaller populations. Data between 1968 and 1978 were available only from Victoria (Dr Rosemary Lester, Acting Director, Disease Control and Research, Public Health Branch, Department of Human Services, VIC, personal communication) because meningococcal disease was not notifiable nationally during these years.7,8 Notifications increased steeply in Queensland in the 1960s and in South Australia in the mid 1980s. However, the former included meningitis caused by coxsackievirus, echovirus, and adenovirus,28 and the latter included genital isolates of Neisseria meningitidis not associated with invasive disease (Dr Scott Cameron, formerly Head, Communicable Diseases Control Unit, South Australian Health Commission, Adelaide, SA, personal communication). The corrected rates in both states for these periods were consistently below 0.7/100 000 population. |
2 Annual notification rates and deaths from meningococcal disease for all states/territories in Australia, 1915–2003
The annual number of deaths from meningococcal disease was obtained from the compilation of mortality data collected by the Australian Bureau of Statistics.29 |
Competing interests
None identified.
Acknowledgements
Christine Phillips provided invaluable comments on earlier drafts and revisions of the manuscript. I am indebted to Rosemary Lester, Scott Cameron and Rob Condon, who shared unpublished data cited in the manuscript.
References
- Australian Government Department of Health and Ageing. Federal government approves free national vaccine program to combat meningococcal disease. Media release. 20 August 2002. http://www.health.gov.au/internet/wcms/publishing.nsf/content/health-mediarel-yr2002-kp-kp02077.htm (accessed Aug 2006).
- Welte R, van den Dobbelsteen G, Bos JM, et al. Economic evaluation of meningococcal serogroup C conjugate vaccination programmes in The Netherlands and its impact on decision-making. Vaccine 2004; 23: 470-479. 0_i1092244
- Snape MD, Pollard AJ. Meningococcal polysaccharide-protein conjugate vaccines. Lancet Infect Dis 2005; 5: 21-30. 0_i1092246
- Butler JC. Nature abhors vacuum, but public health is loving it: the sustained decrease in the rate of invasive Haemophilus influenzae disease. Clin Infect Dis 2005; 40: 831-833. 0_i1092248
- Musher DM. Pneumococcal vaccine — direct and indirect (“herd”) effects. N Engl J Med 2006; 354: 1522-1524. 0_i1092250
- Cumpston JHL. Cerebro-spinal meningitis. In: Lewis MJ, editor. Health and disease in Australia: a history. Canberra: AGPS, 1989: 321-323. 0_i1092252
- Hall R. Notifiable diseases surveillance, 1917 to 1991 data. http://www.health.gov.au/internet/wcms/publishing.nsf/Content/cda-pubs-annlrpt-oz_dis19_91.htm (accessed Aug 2006).
- Hall R. Notifiable diseases surveillance, 1917 to 1991. Commun Dis Intell 1993; 17: 226-236. 0_i1092256
- Clements DA, Gilbert GL. Increase in admissions for Neisseria meningitidis infection in Australia. Lancet 1989; 2: 1464. 0_i1092258
- Hansman D. Epidemiology of meningococcal disease in Australia and New Zealand with a note on Papua New Guinea. In: Vedros N, editor. Evolution of meningococcal disease. Boca Raton, Fl: CRC Press, 1987: 9-18. 0_i1092260
- Hansman D, Ashton F. Serotype and serosubtype distribution of strains of Neisseria meningitidis isolated in South Australia and the Northern Territory of Australia: 1971-1989. Pathology 1994; 26: 318-320. 0_i1092262
- Munro R, Dorman D, Daley D, Tomlinson P. Meningococcal serogroups in New South Wales, 1977-1987. Med J Aust 1988; 149: 360-362. 0_i1092264
- National Neisseria Network. Meningococcal isolate surveillance, Australia, 1994. Commun Dis Intell 1995; 19: 286-289. 0_i1092266
- National Neisseria Network. Meningococcal isolate surveillance, Australia, 1995. Commun Dis Intell 1996; 20: 422-424. 0_pgfId-1092268
- Australian Meningococcal Surveillance Programme. Annual report of the Australian Meningococcal Surveillance Programme 1996. Commun Dis Intell 1997; 21: 217-221. 0_pgfId-1092269
- Australian Meningococcal Surveillance Programme. Annual report of the Australian Meningococcal Surveillance Programme, 1997. Commun Dis Intell 1998; 22: 205-211. 0_pgfId-1092270
- Australian Meningococcal Surveillance Programme. Annual report of the Australian Meningococcal Surveillance Programme, 1998. Commun Dis Intell 1999; 23: 317-323. 0_i1092271
- Australian Meningococcal Surveillance Programme. Annual report of the Australian Meningococcal Surveillance Programme, 1999. Commun Dis Intell 2000; 24: 181-189. 0_pgfId-1092273
- Australian Meningococcal Surveillance Programme. Annual report of the Australian Meningococcal Surveillance Programme, 2000. Commun Dis Intell 2001; 25: 113-121. 0_pgfId-1092274
- Australian Meningococcal Surveillance Programme. Annual report of the Australian Meningococcal Surveillance Programme, 2001. Commun Dis Intell 2002; 26: 407-418. 0_pgfId-1092275
- Australian Meningococcal Surveillance Programme. Annual report of the Australian Meningococcal Surveillance Programme, 2002. Commun Dis Intell 2003; 27: 196-208. 0_i1092276
- Australian Meningococcal Surveillance Programme. Annual report of the Australian Meningococcal Surveillance Programme, 2003. Commun Dis Intell 2004; 28: 194-206. 0_i1092278
- Achtman M. Clonal spread of serogroup A meningococci: a paradigm for the analysis of microevolution in bacteria. Mol Microbiol 1994; 11: 15-22. 0_i1092280
- Caugant DA. Population genetics and molecular epidemiology of Neisseria meningitidis. APMIS 1998; 106: 505-525. 0_i1092282
- Olyhoek T, Crowe BA, Achtman M. Clonal population structure of Neisseria meningitidis serogroup A isolated from epidemics and pandemics between 1915 and 1983. Rev Infect Dis 1987; 9: 665-692. 0_i1092284
- Schwartz B, Moore PS, Broome CV. Global epidemiology of meningococcal disease. Clin Microbiol Rev 1989; 2 Suppl: S118-S124. 0_i1092286
- Australian Government Department of Health and Ageing. National notifiable diseases surveillance system [database]. http://www9.health.gov.au/cda/source/Rpt_2_sel.cfm (accessed Aug 2005).
- Health and Medical Services Branch. Annual reports on the health and medical services of the state of Queensland. Brisbane: Queensland Government, 1945-1983. 0_i1092290
- Australian Institute of Health and Welfare. General record of incidence of mortality books. Version 8. Meningococcal infection (ICD-10 A39). Canberra: AIHW, 2005. 0_i1092292
- Holmes MJ. Report on cerebro-spinal meningitis. Med J Aust 1941; 1: 541-548. 0_i1092294
- Peltola H. Meningococcal disease: still with us. Rev Infect Dis 1983; 5: 71-91. 0_i1092296
- Creasey SA. Epidemic meningococcal meningitis in central Australia in the 1970s. Med J Aust 1991; 155: 725-726. 0_i1092298
- Patel MS, Merianos A, Hanna JN, et al. Epidemic meningococcal meningitis in central Australia, 1987–1991. Med J Aust 1993; 158: 336-340. 0_i1092300
- Harrison L, Broome C. The epidemiology of meningococcal meningitis in the US civilian population. In: Vedros N, ed. Evolution of meningococcal disease. Boca Raton, Fl: CRC Press, 1987: 28-45. 0_i1092302
- Jones DM. Epidemiology of meningococcal infection in England and Wales. J Med Microbiol 1988; 26: 165-168. 0_i1092304
- Heyderman RS, Ben-Shlomo Y, Brennan CA, Somerset M. The incidence and mortality for meningococcal disease associated with area deprivation: an ecological study of hospital episode statistics. Arch Dis Child 2004; 89: 1064-1068. 0_i1092306
- Connolly M, Noah N. Is group C meningococcal disease increasing in Europe? A report of surveillance of meningococcal infection in Europe 1993-6. European Meningitis Surveillance Group. Epidemiol Infect 1999; 122: 41-49. 0_i1092308
- Counts GW, Gregory DF, Spearman JG, et al. Group A meningococcal disease in the US Pacific Northwest: epidemiology, clinical features, and effect of a vaccination control program. Rev Infect Dis 1984; 6: 640-648. 0_i1092310
- Lennon D, Voss L, Sinclair J, Heffernan H. An outbreak of meningococcal disease in Auckland, New Zealand. Pediatr Infect Dis J 1989; 8: 11-15. 0_i1092312
- Patel M. Meningococcal disease in Australia: looking at the past, thinking of the future. Commun Dis Intell 1997; 21: 233-236. 0_i1092314
- Jelfs J, Munro R, Ellis J. PFGE-RFLP analysis of meningococci of the phenotype C:2b:P1.2 causing geographically diverse outbreaks of disease in Australia. Tenth International Pathogenic Neisseria Conference; 1996 Sep 8-13; Baltimore, Md: 467-468. 0_i1092316
- Munro R, Kociuba K, Jelfs J, et al. Meningococcal disease in urban south western Sydney, 1990-1994. Aust N Z J Med 1996; 26: 526-532. 0_i1092318
- Jelfs J, Jalaludin B, Munro R, et al. A cluster of meningococcal disease in western Sydney, Australia initially associated with a nightclub. Epidemiol Infect 1998; 120: 263-270. 0_i1092320
- Robinson P, Griffith J, Taylor K, et al. Laboratory enhanced surveillance for meningococcal disease in Victoria. J Paediatr Child Health 2001; 37: S7-S12. 0_i1092322
- Baker MG, Martin DR, Kieft CE, Lennon D. A 10-year serogroup B meningococcal disease epidemic in New Zealand: descriptive epidemiology, 1991-2000. J Paediatr Child Health 2001; 37: S13-S19. 0_i1092324
- Meyers LA, Levin BR, Richardson AR, Stojiljkovic I. Epidemiology, hypermutation, within-host evolution and the virulence of Neisseria meningitidis. Proc Biol Sci 2003; 270: 1667-1677. 0_i1092326
- Maiden M. Population structure of Neisseria meningitis. In: Ferreiros C, Criado MT, Vazquez J, editors. Emerging strategies in the fight against meningitis: molecular and cellular aspects. Norwich, UK: Horizon Scientific Press, 2002: 151-170. 0_i1092328
- Baker M, McNicholas A, Garrett N, et al. Household crowding a major risk factor for epidemic meningococcal disease in Auckland children. Pediatr Infect Dis J 2000; 19: 983-990. 0_i1092330
- Reid G. When microbe meets human. Clin Infect Dis 2004; 39: 827-830. 0_i1092332
- Patel M, Lee CK. Polysaccharide vaccines for preventing serogroup A meningococcal meningitis (Cochrane review). The Cochrane Library, Issue 1, 2005 (CD001093). Chichester, UK: John Wiley & Sons, Ltd. 0_i1092334
- Rosenstein N, Levine O, Taylor JP, et al. Efficacy of meningococcal vaccine and barriers to vaccination. JAMA 1998; 279: 435-439. 0_i1092336
- Gold R. Epidemiology of meningococcal disease in light of recent Hajj-associated outbreaks. Clin Infect Dis 2003; 36: 684-686. 0_i1092338
- Pelton SI. Modifying acute otitis media: implications of immunization with pneumococcal conjugate vaccine. Pediatr Infect Dis J 2004; 23: 839-841. 0_i1092340
- Esposito S, Cavagna R, Bosis S, et al. Emerging role of Mycoplasma pneumoniae in children with acute pharyngitis. Eur J Clin Microbiol Infect Dis 2002; 21: 607-610. 0_i1092342
- Goldschneider I, Gotschlich EC, Artenstein MS. Human immunity to the meningococcus. II. Development of natural immunity. J Exp Med 1969; 129: 1327-1348. 0_i1092344
- Maiden MC, Spratt BG. Meningococcal conjugate vaccines: new opportunities and new challenges. Lancet 1999; 354: 615-616. 0_i1092347