





Volume 185 - Issue 7
Alzheimer’s disease and frontotemporal dementia: prospects of a tailored therapy?
Authors: Jürgen Götz, Lars M Ittner and Nicole Schonrock
Med J Aust 2006; 185 (7): 381-384. || doi: 10.5694/j.1326-5377.2006.tb00615.x
Published online: 2 October 2006
Published online: 2 October 2006
Abstract
Alzheimer’s disease (AD) is the most prevalent dementia (accounting for 50%–75% of cases of dementia in people aged over 65 years), followed by frontotemporal dementia (FTD) (10%–20% of cases).
AD is characterised histopathologically by Aβ-containing amyloid plaques and tau-containing neurofibrillary tangles, whereas FTD exhibits neurofibrillary tangles alone.
Current symptomatic treatments of AD are of limited benefit, as they are not directed at the underlying biological basis of the disease.
The development of transgenic animal models has provided insight into disease mechanisms and helped define novel drug targets.
More than 50 drugs are currently in clinical trials, and novel and more effective drugs targeting both AD and FTD are expected to become available within 5–10 years.
Dementia is defined as a chronic or progressive loss of cortical and subcortical functions resulting in a complex cognitive decline. This is commonly accompanied by disturbances of mood, behaviour and personality. Because of increasing life expectancy, neurodegenerative dementias are becoming more common. In developed countries, the prevalence of dementia is 1.5% among 65-year-olds, doubling with every 4 years of increasing age to reach 30% among 80-year-olds.1 Alzheimer’s disease (AD) is the most prevalent of the dementias (accounting for 50%–75% of cases), followed by frontotemporal dementia (FTD) (10%–20%), vascular dementia (10%–15%) and dementia with Lewy bodies (10%–15%).2 The social and economic burden associated with these dementias is enormous. Currently, 160 000 Australians are affected, but with the ageing of the population, this will increase to an estimated 500 000 by 2040. The annual cost to the Australian economy is about $6.5 billion, corresponding to 1% of the gross domestic product, and is estimated to reach 3.3% by the middle of this century.3
Limitations of current treatment
The current symptomatic treatment of AD is of limited benefit, as it is not directed at the underlying biological basis of the disease. Treatments include the acetylcholine esterase inhibitors (eg, donepezil hydrochloride, galantamine hydrobromide and rivastigmine tartrate), which stabilise the neurotransmitter acetylcholine in the synaptic cleft, and the N-methyl-D-aspartate receptor antagonist, memantine, which counteracts the deleterious effects of high brain concentrations of the excitatory amino acid, glutamate. Unfortunately, these drugs can have severe psychotropic side effects in patients with FTD.4 Treatment in general is further complicated by the fact that FTD is very heterogeneous, comprising several clinical entities.5,6
Biological basis of AD and FTD
Histopathologically, both AD and FTD are characterised by insoluble filamentous aggregates in the brain (Box 1). They share this feature with Parkinson’s disease and Creutzfeldt–Jakob disease. The biological basis of AD and FTD is discussed in Box 2. As both diseases involve lesions containing the microtubule-associated protein tau, they are classed as tauopathies (see Box 3 for definitions). AD is characterised histopathologically by Aβ-containing amyloid plaques and tau-containing neurofibrillary tangles, whereas FTD exhibits neurofibrillary tangles alone.
Clinical features
The clinical presentation of AD is dominated by early memory deficits, followed by gradual erosion of other cognitive functions such as judgement, verbal fluency or orientation. Memory impairment is usually the first and dominating feature and reflects the fact that the most severe neuropathological changes occur in the hippocampal formation, followed by the association cortices and subcortical structures.
FTD is the preferred term for a spectrum of dementias characterised by focal atrophy of frontal and anterior temporal regions. FTD is mainly associated with behavioural impairment (such as unrestrained behaviour resulting from a lessening or loss of inhibitions and personal and social awareness), followed by affective symptoms, speech disorder and memory problems. In many cases, additional degenerative changes are observed in subcortical brain regions, such as the substantia nigra, leading to Parkinsonian symptoms.16-18 One category of FTD lacks tau aggregates; this includes FTD with motor neurone inclusions (tau-negative and ubiquitin-positive inclusions).17,18 In exciting work published this year, mutations were identified in affected families in the progranulin gene (PGRN).19 PGRN has also been strongly linked to tumorigenesis. Moreover, PGRN expression is increased in activated microglia in many neurodegenerative diseases, including Creutzfeldt–Jakob disease, motor neurone disease and AD. The identity of the ubiquitinated protein in the inclusions is not known.19,20
AD and FTD have a distinct neuropathological profile but, as outlined above, mixed states are probably more frequent than pure dementia syndromes.1 Also, in clinical practice, AD and FTD are sometimes difficult to differentiate.
Transgenic animal models
During the past 15 years, transgenic mice have evolved as the preferred model for AD research. Expression of the amyloid precursor protein carrying mutations found in familial AD cases (see Box 2, Figure) has been found to cause Aβ plaque formation and concomitant memory deficits that progress with age; however, neurofibrillary tangle formation has not been reproduced (reviewed by Götz et al9). Similarly, tau transgenic models have been developed, with the first strain being established by Götz et al in 1995, showing aggregation of hyperphosphorylated tau,21 followed by more advanced transgenic lines modelling formation of neurofibrillary tangles, but not plaques.22-24 The tau transgenic mice also displayed memory impairment that correlated with the expression pattern of the transgene (reviewed by Götz et al9).
These models have been used to test a range of hypotheses to explain how plaques and neurofibrillary tangles interact and how they impair neuronal functions and eventually lead to massive neuronal degeneration (Box 4). For example, by combining the plaque and the neurofibrillary tangle pathology in double transgenic mice, and by intracerebral injection of Aβ into tau transgenic mice, the amyloid cascade hypothesis has been strengthened.24,25 Impaired axonal transport was found in amyloid precursor protein and tau transgenic mice, and subsequently in early, but not late, stages of AD.26 A mitochondrial dysfunction, as well as an upregulation of antioxidant enzymes in response to oxidative stress, was found in tau transgenic mice.27 Finally, cell cycle re-entry and cell death were reported in wild-type tau transgenic mice, supporting the mitosis failure hypothesis.28
Potential drug targets and clinical trials
More than 50 drugs are now in clinical trials.29 These can be divided into several categories.30
Drugs targeting Aβ pathology: To combat Aβ pathology, early efforts concentrated on inhibiting β- and γ-secretase, the enzymes that release toxic Aβ from amyloid precursor protein.6 A major problem associated with γ-secretase is that it has a range of physiological substrates potentially causing toxic side effects. Indirect inhibition of γ-secretase may be achieved using inhibitors of glycogen synthase kinase-3β (GSK3β; see below). Aβ vaccination (both active and passive) has been shown to be effective in animal models, prompting an active immunisation trial in humans. However, this trial had to be halted because of cases of meningoencephalitis in a subset of patients. Currently, efforts are underway to eliminate side effects using novel active and passive immunisation strategies. Furthermore, statins are used, based on epidemiological studies suggesting that high cholesterol levels have a role in AD pathogenesis. The metal-chelating compound clioquinol has been shown in a phase II trial to decrease Aβ levels in AD patients. Compounds are in phase III trials that have been designed to interfere with the association between glycosaminoglycans and Aβ, and thus to block Aβ fibril formation.
Drugs targeting tau pathology: To combat tau pathology, inhibitors of tau phosphorylation and filament formation are attractive targets. In diseased brain, only a subset of phosphoepitopes of tau are pathological epitopes.31 We have shown two of these to be associated with the Aβ-mediated induction of neurofibrillary tangles.22-24 Many sites are also phosphorylated under physiological conditions, albeit to a lesser extent. Of the kinases regulating tau phosphorylation, GSK3 is a prime suspect. Several GSK3 inhibitors are in development, with sodium valproate in phase III trials as therapy for agitation in AD patients. Another candidate drug is CEP-1347, an inhibitor of stress kinase activation, which is in clinical trials for Parkinson’s disease. As tau aggregation is accompanied by microtubule disassembly, drugs that stabilise microtubules, such as the anti-cancer drug paclitaxel, are currently being evaluated.
Drugs targeting oxidation and inflammation: Finally, some drugs target mechanisms underlying both Aβ- and tau-mediated toxicity. These include drugs aimed at detoxifying reactive oxygen species: vitamin E (α-tocopherol) is undergoing trials for mild cognitive impairment, and various antioxidants for mild cognitive impairment and AD. Non-steroidal anti-inflammatory drugs (NSAIDs) may help prevent or delay the onset of AD by decreasing brain inflammation. These include ibuprofen, naproxen and rofecoxib, which are in phase III trials, but in general do not seem to delay functional decline in AD patients.32
Conclusions
The identification of pathogenic mutations in AD and FTD has greatly increased our understanding of these dementias and paved the way for the development of animal and tissue culture models. These have been invaluable tools in dissecting pathogenic mechanisms and identifying drug targets. It is hoped that a biochemical dissection of disease mechanisms will ultimately lead to tailored treatment strategies, and that new drugs will enter the market within the next 5–10 years to effectively treat patients with AD and FTD.
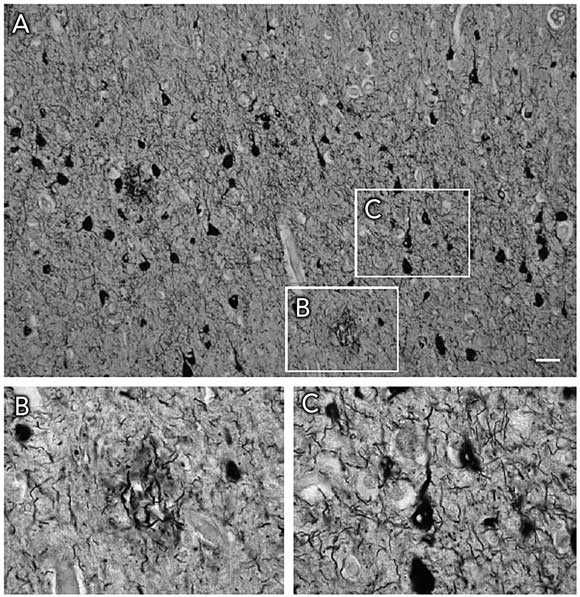
1 Hallmark lesions of Alzheimer’s disease and frontotemporal dementia
A: Temporal lobe of the brain in Alzheimer’s disease, revealing neurofibrillary tangles and neuritic plaques (Gallyas silver stain; scale bar, 12.5 μm). B: Neuritic plaque at higher magnification. C: Neurofibrillary tangle with sparing of the nucleus (centre) at higher magnification. Amyloid plaques are virtually absent in frontotemporal dementia, which thus can be considered a tau-only tauopathy.
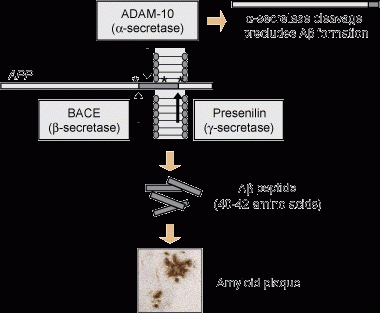
2 Histopathology and genetic basis of Alzheimer’s disease and frontotemporal dementia
Cleavage pathways for amyloid precursor protein (APP)
APP may be cleaved by membrane-associated α-secretase (a disintegrin and metalloproteinase [ADAM] 10), which cleaves it within the Aβ domain, thus precluding formation of Aβ peptide. This pathway is therefore non-amyloidogenic.
Alternatively, APP may be cleaved through the endosomal–lysosomal pathway, first by β-secretase and then by γ-secretase, generating the Aβ peptide. Mutations found in familial cases of Alzheimer’s disease are indicated by asterisks.
Alzheimer’s disease (AD) was initially defined by the presence of extracellular amyloid plaques and intraneuronal neurofibrillary tangles (Box 1).
Amyloid plaques: The major component of amyloid plaques is Aβ, an aggregated peptide comprising 40–42 amino acids (Aβ40 and Aβ42). This peptide is derived by proteolytic cleavage from the larger amyloid precursor protein. The precursor protein is cleaved first by β-secretase and then by γ-secretase, which together generate the Aβ peptide. β-Secretase activity has been attributed to a single protein, BACE (β-site amyloid precursor protein-cleaving enzyme), whereas γ-secretase activity was shown to depend on four components: presenilin, nicastrin, APH-1 (anterior pharynx defective 1) and PEN-2 (presenilin enhancer 2) (Figure). Alternatively, amyloid precursor protein can be proteolytically cleaved by α-secretase, which cleaves it within the Aβ domain. This pathway is non-amyloidogenic (ie, it does not generate the toxic Aβ species6-8).
Neurofibrillary lesions: These lesions are the second histopathological hallmark of AD. They are found in cell bodies and apical dendrites as neurofibrillary tangles, in distal dendrites as neuropil threads, and in the abnormal neurites associated with some Aβ plaques (neuritic plaques). The major components of these lesions are abnormal filaments of the microtubule-associated protein tau. In the course of AD, tau becomes abnormally phosphorylated, adopts an altered conformation and is relocalised from axonal to somatodendritic compartments. Phosphorylation tends to dissociate tau from microtubules. As this increases the soluble pool of tau, it might be an important first step in the assembly of tau filaments. Furthermore, microtubules disintegrate in the course of this process, thereby severely compromising the transport machinery of the neurones.7-9 Evidence is increasing that toxicity is not restricted to the fibrillar species, but that oligomeric forms of tau and Aβ may be even more toxic.
Lewy bodies: In more than 50% of AD cases, neurofibrillary tangles and plaques coexist with a third type of protein deposit, Lewy bodies.10 The latter are the hallmark lesions of Parkinson’s disease, where they are composed of aggregated α-synuclein. In comparison, the Lewy body variant of AD is defined by the presence of all three protein aggregates — Aβ, tau and α-synuclein. These share “amyloid” properties: they have a β-pleated sheet structure and can be stained with dyes such as thioflavin-S.11 Hence, the term “brain amyloidosis” has been coined.
Tauopathies: Tau-containing lesions are found in about two dozen diseases collectively termed tauopathies, with AD being the most common subtype.7-9 However, unlike AD, most tauopathies are characterised by the absence of plaques.12 This has important implications, as an Aβ-directed therapy will likely be ineffective unless it targets common pathogenic mechanisms or common “amyloid” characteristics of the aggregates. In AD, tau aggregates are confined to neurones, but in corticobasal degeneration and progressive supranuclear palsy, they are also abundant in glial cells, such as astrocytes and oligodendrocytes. In Pick’s disease, a specific form of frontotemporal dementia, tau aggregates are found in spherical lesions termed Pick’s bodies. Further differences are in the tau species found in the aggregates, the degree and sites of tau phosphorylation, and the affected brain areas.1 Given the pronounced heterogeneity of frontotemporal dementia and high frequency of mixed types, drug screening efforts should not be restricted to Aβ, but should also target tau.
In the rare familial forms of AD, pathogenic mutations have been identified in both the gene encoding the Aβ precursor (amyloid precursor protein) and in the presenilin genes that encode part of the protease complex involved in Aβ formation13 (Figure). As carriers of these mutations inevitably develop both plaques and neurofibrillary tangles, this genetic evidence has led to the amyloid cascade hypothesis, which claims that, at least in familial forms of AD, Aβ induces neurofibrillary tangles.
For the more frequent forms of late-onset sporadic AD, two dozen risk-conferring genes have so far been identified, including (very recently) ubiquilin,14 but only the apolipoprotein E gene has been confirmed unanimously.15 When sporadic AD is compared with familial AD, the histopathological hallmarks are indistinguishable. This implies that lessons learned from familial AD may also apply to sporadic AD, in particular those derived from animal models expressing familial AD mutant genes.
No mutations have been identified in the tau gene in AD. However, tau mutations have been identified in more than 100 families with familial forms of frontotemporal dementia (FTDP-17), proving that tau dysfunction in itself can lead to neurodegeneration and dementia.
3 Terminology defined
Aβ plaques: Extracellular aggregates mainly composed of highly fibrillogenic 40–42 amino acid polypeptides, which are derived by proteolytic cleavage from the larger amyloid precursor protein.
Frontotemporal dementia (FTD): Category of non-Alzheimer dementias including progressive supranuclear palsy, corticobasal degeneration, FTD with Parkinsonism linked to chromosome 17, and Pick’s disease.
Lewy bodies: Major type of lesion found in Parkinson’s disease, composed of aggregated forms of the protein α-synuclein.
Neurofibrillary tangles: Insoluble filamentous aggregates of hyperphosphorylated tau eventually filling the entire soma of a dying neurone.
α-Synuclein: Major proteinaceous constituent of the Lewy bodies of Parkinson’s disease that shares many features with tau.
Tau: A microtubule-associated protein critical for assembly and maintenance of microtubules in axons; it contains an unusually high number of phosphorylated amino acids.
Tau hyperphosphorylation: Post-translational modification of tau, with physiological epitopes of tau phosphorylated at a higher percentage, and pathological epitopes being phosphorylated de novo.
Transgenic mouse: A mouse carrying a foreign gene (eg, human tau targeted to neurones) introduced by pronuclear injection of oocytes.
4 Hypotheses linking Aβ and tau to neuronal cell death (ie, dementia) and potential therapeutic strategies
Amyloid cascade hypothesis: Drugs targeting enzymes involved in Aβ production (β- and γ-secretase) and clearance (insulin-degrading enzyme, neprilysin), and aggregation blockers/helix breakers.
Tau phosphorylation hypothesis: Drugs targeting kinases, phosphatases, and proteases.
Mitosis failure hypothesis: Drugs targeting cyclins and other cell-cycle regulator proteins.
Oxidative stress hypothesis: Antioxidants.
Transport failure hypothesis: Microtubule-stabilising agents.
Competing interests
None identified.
Acknowledgements
Jürgen Götz is a Medical Foundation Fellow and has been supported by the University of Sydney, the Australian Research Council, the New South Wales Government through the NSW Office for Science and Medical Research (BioFirst program), the National Health and Medical Research Council and the Medical Foundation (University of Sydney). This article is dedicated to Professor Max Bennett (University of Sydney), acknowledging his generous support.
References
- Ritchie K, Lovestone S. The dementias. Lancet 2002; 360: 1759-1766. 0_i1091933
- Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology 2002; 58: 1615-1621. 0_i1091935
- Alzheimer’s Australia. Types of dementia. http://www.alzheimers.org.au (accessed Aug 2006).
- Cummings JL, Askin-Edgar S. Evidence for psychotropic effects of acetylcholinesterase inhibitors. CNS Drugs 2000; 13: 385-395. 0_i1091939
- Allain H, Bentue-Ferrer D, Tribut O, et al. Drug therapy of frontotemporal dementia. Hum Psychopharmacol 2003; 18: 221-225. 0_i1091941
- Small DH. Acetylcholinesterase inhibitors for the treatment of dementia in Alzheimer’s disease: do we need new inhibitors? Expert Opin Emerg Drugs 2005; 10: 817-825. 0_i1091943
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci 2001; 24: 1121-1159. 0_i1091945
- Götz J. Tau and transgenic animal models. Brain Res Brain Res Rev 2001; 35: 266-286. 0_i1091947
- Götz J, Streffer JR, David D, et al. Transgenic animal models of Alzheimer’s disease and related disorders: histopathology, behavior and therapy. Mol Psychiatry 2004; 9: 664-683. 0_i1091949
- Brown DF, Dababo MA, Bigio EH, et al. Neuropathologic evidence that the Lewy body variant of Alzheimer disease represents coexistence of Alzheimer disease and idiopathic Parkinson disease. J Neuropathol Exp Neurol 1998; 57: 39-46. 0_i1091951
- Kurosinski P, Guggisberg M, Götz J. Alzheimer’s and Parkinson’s disease – overlapping or synergistic pathologies? Trends Mol Med 2002; 8: 3-5. 0_i1091953
- Shahani N, Brandt R. Functions and malfunctions of the tau proteins. Cell Mol Life Sci 2002; 59: 1668-1680. 0_i1091955
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 2001; 81: 741-766. 0_i1091957
- Bertram L, Hiltunen M, Parkinson M, et al. Family-based association between Alzheimer's disease and variants in UBQLN1. N Engl J Med 2005; 352: 884-894. 0_i1091959
- Rocchi A, Pellegrini S, Siciliano G, Murri L. Causative and susceptibility genes for Alzheimer’s disease: a review. Brain Res Bull 2003; 61: 1-24. 0_i1091961
- Wszolek ZK, Pfeiffer RF, Bhatt MH, et al. Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Ann Neurol 1992; 32: 312-320. 0_i1091963
- Hodges JR, Davies R, Xuereb J, et al. Survival in frontotemporal dementia. Neurology 2003; 61: 349-354. 0_i1091965
- Hodges JR, Davies RR, Xuereb JH, et al. Clinicopathological correlates in frontotemporal dementia. Ann Neurol 2004; 56: 399-406. 0_i1091967
- Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006; 442: 916-919. 0_i1091969
- Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006; 442: 920-924. 0_i1091971
- Götz J, Probst A, Spillantini MG, et al. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. Embo J 1995; 14: 1304-1313. 0_i1091973
- Lewis J, McGowan E, Rockwood J, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 2000; 25: 402-405. 0_i1091975
- Götz J, Chen F, Barmettler R, Nitsch RM. Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem 2001; 276: 529-534. 0_pgfId-1099100
- Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Abeta 42 fibrils. Science 2001; 293: 1491-1495. 0_i1091978
- Lewis J, Dickson DW, Lin W-L, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001; 293: 1487-1491. 0_i1091980
- Stokin GB, Lillo C, Falzone TL, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 2005; 307: 1282-1288. 0_i1091982
- David DC, Hauptmann S, Scherping I, et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem 2005; 280: 23802-23814. 0_i1091984
- Andorfer C, Acker CM, Kress Y, et al. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci 2005; 25: 5446-5454. 0_i1091986
- Alzheimer Research Forum. Drugs in clinical trials. http://www.alzforum.org (accessed Aug 2006).
- Rosenberg RN. Translational research on the way to effective therapy for Alzheimer disease. Arch Gen Psychiatry 2005; 62: 1186-1192. 0_i1091990
- Chen F, David D, Ferrari A, Götz J. Posttranslational modifications of tau — role in human tauopathies and modeling in transgenic animals. Curr Drug Targets 2004; 5: 503-515. 0_i1091992
- Aisen PS, Schafer KA, Grundman M, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 2003; 289: 2819-2826. 0_i1091994