





Volume 185 - Issue 5
Inhaled corticosteroids, adrenal suppression and benign intracranial hypertension
Authors: Patrick Patradoon-Ho, Hasantha Gunasekera, Monique M Ryan and Geoffrey R Ambler
Med J Aust 2006; 185 (5): 279-280. || doi: 10.5694/j.1326-5377.2006.tb00561.x
Published online: 4 September 2006
Published online: 4 September 2006
Clinical record
A 15-year-old boy developed recurrent, severe occipital headache, with vomiting, lethargy and blurred vision, after a minor head injury without loss of consciousness. The headache was dull and tense, exacerbated by activity and ameliorated by sleep, but unaffected by a change of posture. A week after the head injury, his general practitioner found him to be neurologically normal. The results of a non-contrast cerebral computed tomography scan were also normal.
Based on a history of reversible wheeze and cough, the patient had been diagnosed with asthma at 12 years of age. He was strictly compliant with his prescription of inhaled fluticasone propionate (1000 μg/day via accuhaler) and rinsed his mouth after use. As instructed, he doubled the dose of inhaled corticosteroids during acute attacks, which occurred twice a year. There were no interval symptoms. Nonetheless, his dose of inhaled corticosteroids was never reduced. Before being prescribed inhaled corticosteroids, he had been prescribed nasal steroids for allergic rhinitis and nasal polyps, but rarely required this after a successful surgical intervention. He was not taking any other medications or vitamins. A year before presentation, he had had an episode of witnessed collapse with spontaneous recovery. This was attributed to a vasovagal attack at the time.
Paediatric assessment 1 month after the head injury showed that he was lean and healthy. His weight (49 kg) and height (160.2 cm) were on the 25th centiles, and his body mass index (19 kg/m2) was on the 30th centile. His head circumference (56.8 cm) was on the 75th centile. Neurological examination gave normal results. A Mini-Mental State Examination identified a short-term memory deficit (2/3 objects recalled). The results of a cardiovascular examination were normal except for hypertension (140/70 mmHg; > 97th centile). There were no renal bruits. Urinalysis gave normal results. He was clinically euthyroid, and had no cutaneous hypo- or hyperpigmentation or signs of Cushing syndrome.
He was diagnosed with postconcussion syndrome and reactive hypertension. An ophthalmological assessment identified normal visual fields and acuity and no papilloedema. The fluticasone propionate dose was reduced to 500 μg/day with a view to gradually stopping it altogether.
Over the following 2 weeks, his headache worsened in intensity and frequency. An electroencephalogram and cerebral magnetic resonance scan were normal. Amitriptyline (25 mg/day) was prescribed for the headache, without effect. Two months after the paediatric assessment, lumbar puncture, performed under chloral hydrate sedation when relaxed with the back fully extended, showed an elevated cerebrospinal fluid (CSF) opening pressure of 24 cmH2O (reference range, 10–20 cmH2O).1,2 The procedure resulted in immediate headache relief. Examination of the CSF showed no abnormalities. Benign intracranial hypertension was diagnosed and he was prescribed oral acetazolamide (250 mg four times daily). His inhaled corticosteroid therapy was discontinued.
Persistent mild hypertension (130/70 mmHg; 97th centile) was again noted on repeated occasions. The results of investigations (including renal function, urinalysis, renal ultrasound, full blood count, inflammatory markers, complement factors, antinuclear antibody, electrocardiography, chest x-ray, thyroid function, plasma renin activity, and urinary catecholamines) were normal and did not reveal a secondary cause of the hypertension. However, the morning cortisol level was undetectable (< 6 nmol/L). A morning short Synacthen test (250 μg) confirmed adrenal suppression, with a suboptimal cortisol rise from baseline of 175 nmol/L to a 1-hour peak of 275 nmol/L (reference, > 600 nmol/L). A paired adrenocorticotropic hormone (ACTH) level was 15 pmol/L, not consistent with primary adrenal disease. In view of the adrenal suppression, he was prescribed oral hydrocortisone (20 mg three times daily) for periods of acute physiological stress.
Over the ensuing months, he required multiple therapeutic lumbar punctures, with immediate headache relief on each occasion. The acetazolamide dose was ceased 2 months after initiation. His hypertension resolved 4 months after presentation and the headaches resolved after 9 months. During recovery, he used “stress” hydrocortisone for two episodes of fever and lethargy. Serial (3-monthly) morning short Synacthen tests (250 μg) were performed. By 12 months, he had a normal peak cortisol response (657 nmol/L) and a baseline ACTH level of 7.3 pmol/L. Steroid precautions were then discontinued.
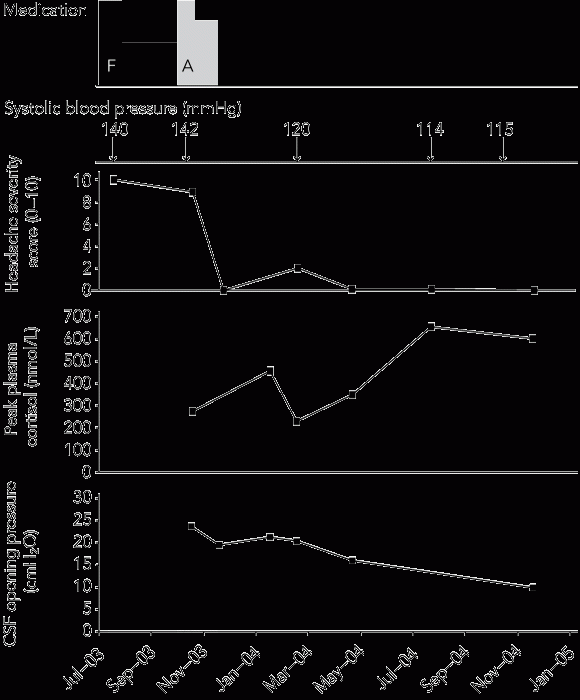
Ophthalmological review now identified a cotton-wool spot on the inferotemporal edge of the right optic disc, suggestive of a past axoplasmic blood flow impediment caused by raised intracranial pressure. The temporal association between his headache severity, CSF opening pressures and cortisol levels is shown in the Box.
Benign intracranial hypertension has an incidence of 1 per 100 000 children.3 As “benign” belies the potentially debilitating nature of this disorder, including the risk of irreversible blindness,4 the synonyms “pseudotumor cerebri” and “idiopathic intracranial hypertension” are sometimes preferred. Although the pathophysiological basis has not been determined, benign intracranial hypertension has been associated with vitamin deficiencies and toxicities, obesity, head injury,5 adrenal insufficiency,6 and medications, including topical and systemic steroids.7-9 To the best of our knowledge, this is the first reported association between benign intracranial hypertension and inhaled corticosteroids (we conducted a MEDLINE search, using multiple search terms, of English language publications from 1966 to June 2005).
The diagnosis of benign intracranial hypertension in our case was based on modified Dandy’s criteria, an elevated cerebrospinal fluid (CSF) opening pressure (using the paediatric reference range),1,2 and repeated instances of immediate headache relief with lumbar puncture. The ophthalmological findings were non-specific but supported the diagnosis. Magnetic resonance venography would have excluded venous sinus thrombosis (an important cause of benign intracranial hypertension), but there was no reason to suspect a hypercoagulable tendency.
As the CSF opening pressure was first measured 2 months after the patient sustained a minor head injury, we cannot determine whether this injury was the trigger for benign intracranial hypertension or incidental to it. However, the injury alone cannot explain the benign intracranial hypertension in this patient, given its mild nature and the absence of any radiological evidence of parenchymal brain injury.
The adrenal suppression was most likely secondary to inhaled corticosteroid therapy. This is supported by the gradual adrenal recovery after withdrawal of inhaled corticosteroids. The past history of collapse suggests the onset of the adrenal suppression may have occurred 1 year before presentation.
Given the absence of papilloedema at presentation, and the fact that the headache and elevated CSF opening pressure were clearly temporally associated with the withdrawal of inhaled corticosteroids, we suggest that the benign intracranial hypertension was related to the withdrawal of the steroid dose. Benign intracranial hypertension has previously only been reported in cases of primary adrenal insufficiency (Addison disease),6 or in association with the use or withdrawal of topical, oral or intranasal corticosteroids.7-9
Clinicians should be mindful of benign intracranial hypertension as a potential side effect when prescribing inhaled corticosteroids. Our case emphasises the importance of using appropriate inhaled corticosteroid dosage and “back titration”. Patients receiving inhaled corticosteroids require regular review. Dosages should be monitored to achieve the minimal dose for symptom control.10
Competing interests
None identified.
References
- Soler D, Cox T, Bullock P, et al. Diagnosis and management of benign intracranial hypertension. Arch Dis Child 1998; 78: 89-94. 0_i1091840
- Friedman DI, Rausch EA. Headache diagnoses in patients with treated idiopathic intracranial hypertension. Neurology 2002; 58: 1551-1553. 0_i1091842
- Gordon K. Pediatric pseudotumor cerebri: descriptive epidemiology. Can J Neurol Sci 1997; 24: 219-221. 0_i1091844
- Wall M. Idiopathic intracranial hypertension. Semin Ophthalmol 1995; 10: 251-259. 0_i1091846
- Dhiravibulya K, Ouvrier R, Johnston I, et al. Benign intracranial hypertension in childhood: a review of 23 patients. J Paediatr Child Health 1991; 27: 304-307. 0_i1091848
- Condulis N, Germain G, Charest N, et al. Pseudotumor cerebri: a presenting manifestation of Addison’s disease. Clin Pediatr (Phila) 1997; 36: 711-713. 0_i1091850
- Grant DN. Benign intracranial hypertension: a review of 79 cases in infancy and childhood. Arch Dis Child 1971; 46: 651-655. 0_i1091852
- Bond DW, Charlton CP, Gregson RM. Drug points: benign intracranial hypertension secondary to nasal fluticasone propionate. BMJ 2001; 322: 897. 0_pgfId-1091854
- Neville BGR, Wilson J. Benign intracranial hypertension following corticosteroid withdrawal in childhood. BMJ 1970; 3: 554-556. 0_i1091856
- van Asperen PP, Mellis CM, Sly PD. The Thoracic Society of Australia and New Zealand. The role of corticosteroids in the management of childhood asthma. Med J Aust 2002; 176: 168-173. 0_i1091858