





Volume 182 - Issue 9
Life-threatening allergic bronchopulmonary aspergillosis in a well child with cystic fibrosis
Authors: Emma Skowronski and Dominic A Fitzgerald
Med J Aust 2005; 182 (9): 482-483. || doi: 10.5694/j.1326-5377.2005.tb06792.x
Published online: 2 May 2005
Published online: 2 May 2005
Allergic bronchopulmonary aspergillosis (ABPA) is an uncommon condition which may complicate asthma and cystic fibrosis; it is seldom considered life-threatening. We report a well 8-year-old boy with cystic fibrosis and normal lung function who progressed to respiratory failure over several days, attributable to ABPA. He recovered with non-invasive ventilation and oral corticosteroid and antifungal medications, regaining normal lung function within 2 months. To our knowledge, such an acute severe presentation of ABPA in a previously well child has not been reported before.
Clinical record
An 8-year-old boy with pancreatic insufficient, homozygous ΔF508 cystic fibrosis (CF) was transferred from a district hospital with a 7-day history of progressive cough, wheeze and tachypnoea, despite 4 days of broad-spectrum antibacterial therapy (intravenous flucloxacillin and cefotaxime with oral roxithromycin), frequent nebulised salbutamol, oral prednisone (2 mg/kg/day) and chest physiotherapy. There were no other systemic symptoms such as rash, myalgia, arthralgia, diarrhoea or headaches. Initial sputum cultures isolated only normal respiratory flora. The provisional diagnosis was an atypical lower respiratory tract infection, attributed to a viral infection or Mycoplasma pneumoniae.
Cystic fibrosis had been diagnosed at newborn screening. His height and weight had tracked along the 10th percentile. Lung function had been normal. Previous sputum cultures had grown Staphylococcus aureus but never Pseudomonas aeruginosa or Burkholderia cepacia. He had not previously wheezed.
On arrival, he was mildly dyspnoeic on supplemental mask oxygen at 6 L/minute with a blood oxygen saturation (Spo2) of 94%. He was tachypnoeic (44 breaths per minute), tachycardic (112 beats per minute), afebrile and normotensive. He had bilateral expiratory wheeze with basal crackles.
Within 24 hours of transfer, our patient’s condition deteriorated, and he required more frequent nebulised salbutamol and developed signs of respiratory fatigue. At this point an arterial blood gas analysis in 12 L of mask oxygen showed: pH, 7.3; partial pressure of oxygen (Po2), 58 mmHg; partial pressure of carbon dioxide (Pco2), 62.7 mmHg; bicarbonate (Hco3) level, 30 mmol/L; and base excess, 2.7. He was transferred to the paediatric intensive care unit (PICU) for respiratory support with mask continuous positive airway pressure (CPAP).
Subcutaneous emphysema developed over the chest wall and neck during the first 48 hours of his PICU admission. A full blood count showed leukocytosis (19.6 × 109/L), and his initial mycoplasma complement fixation test titre was low (< 4). The mycoplasma IgM test result was subsequently negative. Immunofluorescence and culture of nasopharyngeal secretions to isolate a viral pathogen were negative. Total serum IgE titre was 1664 IU (normal range, 0–180 IU) and the skin prick test was positive for Aspergillus fumigatus. The provisional diagnosis was changed to allergic bronchopulmonary aspergillosis (ABPA). Prednisone therapy was continued, and antifungal treatment with oral itraconazole (100 mg twice daily) was added to his therapy.
His respiratory status gradually improved and he was weaned off the nasal mask CPAP after 5 days, avoiding endotracheal intubation. He had clinically apparent subcutaneous emphysema, a small pneumomediastinum, but no pneumothoraces during his PICU stay. The chest radiograph before discharge from PICU (Box 1) showed increased perihilar opacities and left lower lobe infiltrates with resolution of the pneumomediastinum and subcutaneous emphysema. His improving spirometry measurements with treatment are shown in Box 2. He was discharged on Day 13 of admission with marked improvement in symptoms and an FEV1 (forced expiratory volume in 1 second) 66% of predicted.
A provisional diagnosis of ABPA was confirmed by the significantly elevated serum IgE titre, positive skinprick tests for aspergillus, positive IgG aspergillus precipitins (× 4) and clinical findings consistent with the diagnostic criteria outlined in Box 3.1-5 He received decreasing doses of oral corticosteroids over 3.5 months, as well as oral itraconazole. Within 2 months, his spirometry results had returned to normal. Predictably, he became transiently cushingoid, developed mild untreated hypertension (maximum recorded blood pressure, 116/84 mmHg), and gained 4 kg in weight, but had no glycosuria. He was weaned from corticosteroids and itraconazole, and the side effects resolved within 3 months of discontinuing corticosteroids. Ten months later, he remains well, with normal lung function.
Alllergic bronchopulmonary aspergillosis (ABPA) is an uncommon condition that can complicate asthma and cystic fibrosis (CF),6,7 and is seldom considered life-threatening.7 The rapid deterioration in the condition of our previously well patient highlights the spectrum of disease severity that can occur in ABPA, and is a reminder that not all deteriorations in respiratory function in patients with CF are attributable to Pseudomonas aeruginosa or Burkholderia cepacia.7,8 Furthermore, this patient’s prompt clinical response to systemic corticosteroids with a return to normal lung function within 2 months suggests that his prognosis will not be adversely affected.9
The presence of Aspergillus species in the sputum cultures of patients with CF has been reported in up to 57% of patients,9 yet the prevalence of ABPA is reported to be between 2% and 14%.1,6 The diagnostic criteria are listed in Box 3. ABPA is a hypersensitivity reaction to the inhalation of aspergillus spores manifesting as chronic wheeze, pulmonary infiltrates and systemic immune activation.5 This results in elevated IgE, IgG and IgA titres.9 Interestingly, although aspergillus grows in the bronchial mucus, this is not an invasive disease.3-5,9 The exact mechanism by which bronchial wall damage evolves and how this gives rise to bronchiectasis and fibrosis is poorly understood.10
Aspergillus fumigatus infection often occurs months before a clinical diagnosis of ABPA is considered. Moreover, as about 40%–50% of school-aged patients with CF have aspergillus in their sputum, comparatively few develop ABPA.3 The reasons for this are not clear, but presumably relate to genetic predisposition, host defences and environmental exposure to aspergillus. Most children with CF who develop ABPA have relatively mild symptoms, respond to treatment over weeks and can often avoid hospital admission altogether.7 Patients with ABPA complicating CF more commonly follow a course of gradually worsening lung function (because of progression of their CF related bronchiectasis) with recurring relapses of ABPA, particularly in summer and autumn, when spore levels in the environment are at their highest.9,10
Treatment of ABPA has centred on the use of systemic corticosteroids for periods of 2–6 months, reducing from doses of 1.0 mg/kg/day of prednisone.4 The adjunctive use of oral antifungals has been advocated for the treatment of ABPA complicating CF11,12 and asthma.6 The response can be dramatic, as in our patient (Box 2). Itraconazole in combination with inhaled corticosteroids was recently shown to be useful for reducing eosinophilic airway inflammation, reducing systemic immune activation and reducing severe exacerbations over a period of 16 weeks in adults with ABPA complicating asthma.13 The use of itraconazole in children is less well studied. However, a recent case series of patients aged 9 to 33 years with CF and ABPA, treated with inhaled budesonide (800–1600 μg/day) and itraconazole (400–600 mg/day), showed a high prevalence of biochemical adrenal insufficiency on adrenocorticotropin testing. This was attributed to an increased systemic budesonide concentration through a reduced or inhibited metabolism (potentially caused by itraconazole), leading to inhibited steroidogenesis.14 This reminds us to use caution when treating patients who take inhaled steroids with courses of itraconazole for exacerbations of ABPA.
In conclusion, we are unable to find another case report in which a previously well child with ABPA presented with severe acute respiratory failure. This case highlights the importance of considering a diagnosis of ABPA in highly unusual presentations which may complicate cystic fibrosis.
1 Chest x-ray 3 days after admission to intensive care
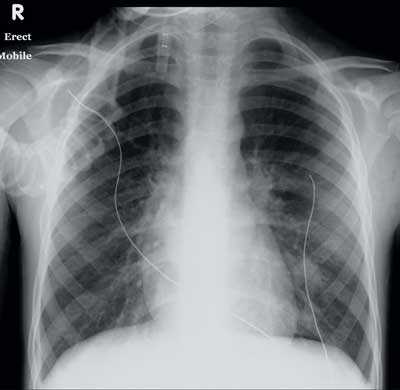
Shows hyperinflated lung fields, perihilar inflammatory changes, emergence of an interstitial infiltrate in the left lower lobe and a small right-sided pleural effusion.
2 Relationship between IgE levels and FEV1 (forced expiratory volume in one second) before, during and after admission
Inverse relationship between IgE levels and FEV1 over time (non-linear scale), showing the drop in lung function at the peak of the disease.
3 Classic case criteria for the diagnosis of allergic bronchopulmonary aspergillosis (ABPA) in patients with cystic fibrosis1-5
Clinical deterioration (increased cough, wheezing, exercise intolerance, increased sputum, decrease in pulmonary function)
Immediate cutaneous reactivity to aspergillus or presence of serum IgE from A. fumigatus
Total serum IgE concentration >1000 IU/L
Precipitating antibodies to A. fumigatus or serum IgG from A. fumigatus
Abnormal chest x-ray (infiltrates, mucus plugging, or a change from earlier films)
Adapted from the ABPA Consensus Conference of the Cystic Fibrosis Foundation4
Acknowledgements
We thank Dr Jonathan Gillis and the intensive care specialists, and Associate Professor Albert Lam for his assistance in providing the chest radiographs for presentation.
References
- Mastella G, Rainisio M, Harms HK, et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis. A European epidemiologic study. Eur Respir J 2000; 16: 464-471.
- Mroueh S, Spock A. Allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. Chest 1994; 105: 32-36.
- Proceedings of the Cystic Fibrosis Foundation ABPA consensus conference. Bethesda, Md: 2001 Jun 12–13. Bethesda, Md: North American Cystic Fibrosis Foundation, 2002. i1085812
- Greenberger PA. Allergic bronchopulmonary aspergillosis. J Allergy Clin Immunol 2002; 110: 685-692. i1085814
- Varkey B. Allergic bronchopulmonary aspergillosis: clinical perspectives. Immunol Allergy Clin North Am 1998; 18: 479-501. i1085816
- Stevens DA, Schwartz HJ, Lee JY, et al. A randomized trial of itraconazole in allergic bronchopulmonary aspergillosis. N Engl J Med 2000; 342: 756-762. i1085818
- Vaughan LM. Allergic bronchopulmonary aspergillosis. Clin Pharm 1993; 12: 24-33. i1085820
- Fitzgerald DA, Cooper DM, Paul M, et al. Burkholderia cepacia in cystic fibrosis: novel Australian cluster strain without accelerated respiratory deterioration. J Paediatr Child Health 2001; 37: 130-136. i1085822
- Lee TM, Greenberger PA, Patterson R, et al. Stage V [fibrotic] allergic bronchopulmonary aspergillosis. A review of 17 cases followed from diagnosis. Arch Intern Med 1987; 147: 319-323. i1085824
- Knutsen AP, Bellone C, Kauffman H. Immunopathogenesis of allergic bronchopulmonary aspergillosis. J Cystic Fibrosis 2002; 2: 76-89. i1085826
- Klein NC, Cunha BA. New antifungal drugs for pulmonary mycoses. Chest 1996; 110: 525-532. i1085828
- Nepomuceno IB, Esrig S, Moss RB. Allergic bronchopulmonary aspergillosis in cystic fibrosis: role of atopy and response to itraconazole. Chest 1999; 115: 364-370. i1085830
- Wark PAB, Hensley MJ, Saltos N, et al. Anti-inflammatory effect of itraconazole in stable allergic bronchopulmonary aspergillosis: a randomized controlled trial. J Allergy Clin Immunol 2003; 111: 952-957. i1085832
- Skov M, Main KM, Sillesen IB, et al. Iatrogenic adrenal insufficiency as a side-effect of combined treatment of itraconazole and budesonide. Eur Respir J 2002; 20: 127-133. i1085834