




In the 50 years since the structure of DNA was elucidated, the human genome has been completely sequenced, and about 30 000 genes have been identified. However, to understand the functions of these genes, attention has now turned to the proteins they encode — the “proteome”. Proteins are responsible for the functional diversity of cells, they perform most biological functions, and it is at their level that many regulatory processes take place, many disease processes occur, and most drug targets are found.
As genomics (the study of genes) has proven inadequate to predict the structure and dynamic properties of all proteins, a new field — proteomics — has developed (Box 1). This is the large-scale study of protein expression, structure and function. It aims to correlate the structural and functional diversity of proteins with underlying biological processes, including disease processes. Proteomics has created opportunities to identify, investigate and target proteins that are differentially expressed in health and disease. Clinical research is poised to benefit enormously from these studies, with the potential to develop better diagnostic and prognostic tests, to identify new therapeutic targets and ultimately to allow patient-individualised therapy.1,2
Here we describe proteomics technology, how it is being used to study disease, and its potential to improve diagnosis and treatment of a range of conditions.
Proteomics was made possible by a number of developments in technology and informatics (Box 2).
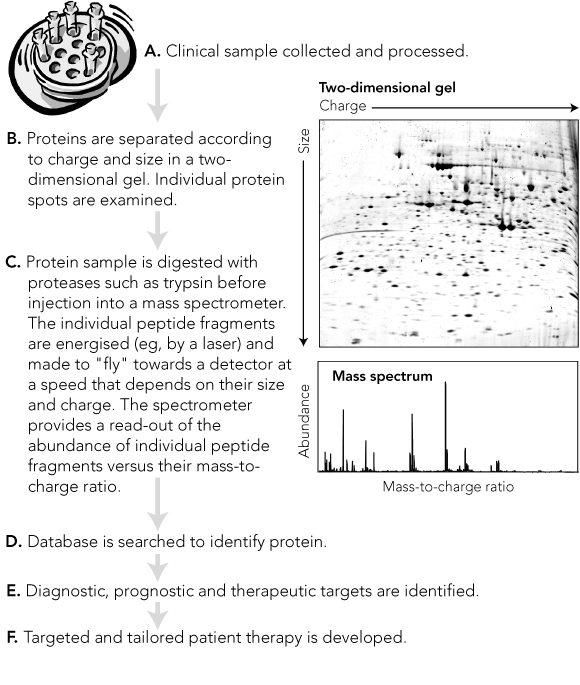
This is a commonly used and highly versatile technique for separating proteins according to their size and charge. Proteins from body fluids, tissues or cells are first separated on a thin gel layer according to their isoelectric charge (first dimension). This gel is then placed across a larger gel, and the proteins are separated in the second dimension, at right angles to the first, according to their size. This technique can separate (resolve) over 1000 proteins per gel. Individual proteins can then be excised and purified for identification, usually by mass spectrometry (see Box 3B).
Mass spectrometry has revolutionised proteomics, allowing thousands of proteins to be analysed rapidly. First developed early in the 20th century, the technique identifies a substance by sorting according to mass, a stream of electrified particles (ions) derived from the substance. The application of mass spectrometry to proteins was made possible by the development of methods for ionising large biomolecules without destroying them. These include electrospray ionisation and matrix-assisted laser desorption ionisation (MALDI).
For mass spectrometry, a protein is digested with an enzyme (usually trypsin), which cleaves it at specific amino acid sequences. The resulting peptide fragments are energised, most often by a laser (eg, using MALDI), but sometimes by electrospray ionisation. The charged peptide fragments are then separated in the spectrometer according to their mass-to-charge ratio, by a method that varies between different types of spectrometers (eg, “time of flight” [TOF] through a flight tube). The separated fragments impinge on a detector, which measures the signal intensity of each fragment.
The graph of this signal intensity against the mass-to-charge ratio is the mass spectrum (Box 3C). It contains a series of peaks, each corresponding to a particular peptide fragment (with a specific amino acid sequence indicated by its mass), and a height representing the relative abundance of that fragment. The peptides in these peaks are then further fragmented to give a second (tandem) mass spectrum, in which each peptide fragment differs from its neighbour by one amino acid. From this, the peptide sequence is determined. The process is termed peptide mass fingerprinting. The peptide sequence is then interrogated against protein databases to identify the original protein.
Continuing developments and improvements in proteomics technology, such as difference-gel 2D-electrophoresis (DIGE; Box 2) and liquid chromatography linked to mass spectrometry, are now allowing proteins to be detected with high sensitivity and specificity in small volumes of biological samples such as blood and urine. Plasma proteins can currently be detected over a concentration range of 10-3 to 10-15. Further development of mass spectrometers, combined with techniques to remove interfering proteins, such as immunodepletion, will enable even lower concentrations to be detected.
In addition, protein microarray systems (“chips”) are being developed as matrix-support surfaces for binding selected proteins in preparation for mass spectrometry. Analogous to DNA chips, these protein chips aim to bind individual proteins from biological samples, such as serum and urine, to allow high-throughput screening for disease-associated proteins.2
The identification and examination of disease markers is currently based on individual proteins, which is not always reliable. For example, assay of prostate serum antigen (PSA) is used to screen for prostate cancer, but levels of this antigen are also raised in benign conditions of the prostate. Advances in proteomics technology allow the simultaneous analysis of thousands of low molecular weight proteins, which may reveal patterns of disease and are potentially useful for early detection and assessing prognosis. The potential therefore exists to use a panel of diagnostic markers to more accurately identify a given disease state.
Most proteomics disease studies have focused on cancer, where proteomics has the potential to allow earlier diagnosis. This is particularly important in ovarian cancer, as most women with this cancer have advanced disease at diagnosis, with a 5-year survival rate of 35%.3 However, as stage I ovarian cancer has a 5-year survival rate of over 90%, early diagnosis is likely to directly affect mortality. A proteomics study has identified serum protein patterns that distinguished patients with ovarian cancer from unaffected women with a positive predictive value of 94%.4 However, the numbers assessed were small, and these results have proven difficult to reproduce.5 This highlights the need to identify the proteins of interest to determine the biological plausibility of the test, and also to undertake prospective population-based assessments to determine the value of proteomic patterns as a screening tool for ovarian cancer. Identifying the proteins involved is also important because of their potential as therapeutic targets.
In cases of cancer in the abdomen, it is occasionally difficult to determine the tissue of origin and consequently the optimal therapy. For example, ovarian and colon cancer can be difficult to distinguish, and correct diagnosis is essential as their treatments vary markedly. Using a combination of genomic and proteomic tools, investigators were able to identify differential markers for the two cancers – the protein villin for colon cancer cells and moesin for ovarian cancer cells.6
Tuberculosis affects millions of people worldwide, and drug-resistant Mycobacterium tuberculosis strains are an increasing problem. A serum screening test that could detect pre-clinical infection would allow early treatment, potentially reducing transmission, and have widespread application. Proteomic techniques have identified proteins secreted in vitro by common clinical isolates. Two of these (rRv3369 and rRv3874) have shown potential as serodiagnostic antigens, with sensitivity of 60%–74% and specificity of 96%–97% in clinical studies.7 These proteins are potential candidates for a kit-based serum screening test.
The pathogenesis of severe acute respiratory syndrome (SARS) is not well understood, and a specific diagnostic method is critical for the management and control of this disease. Proteomic analysis of sera from patients with SARS has identified potential protein markers — truncated forms of α(1)-antitrypsin — which were consistently found in higher concentrations in the sera of SARS patients compared with healthy controls.8 These markers may prove useful as diagnostic tools and therapeutic targets. Moreover, studies of the protein structure of the SARS virus may reveal potential vaccine targets.9
Immune rejection is a major problem after cardiac transplantation. Accurate diagnosis relies on invasive endomyocardial biopsy. A recent study of cardiac biopsy specimens identified over 100 proteins that were upregulated during rejection, including cardiac and heat shock proteins (the latter can be upregulated as a stress response).10 Two of these — αB-crystallin (a heat shock protein) and tropomyosin (a cardiac muscle protein) — were expressed at significantly higher levels in sera of patients undergoing rejection.10 Proteomic analysis of serum may therefore be a powerful and less invasive method of identifying cardiac rejection.
Although still in its infancy, proteomic strategies are also being used to investigate differential protein expression in cancer cells, which may reflect differences in invasiveness and predisposition to developing resistance to treatment. For example, proteomics has identified proteins involved in multidrug resistance (major vault protein) and metastasis (cystatin B) that are highly expressed in glioblastoma multiforme compared with normal brain tissue. These proteins have potential as diagnostic, drug resistance and invasiveness markers for glioblastoma tumors.11 Identifying proteome profiles associated with intrinsic or acquired drug resistance might allow treatment to be varied to avoid this resistance. Hypothetically, it might also be possible to identify tumours that are highly drug-responsive, allowing lower drug doses to be used, potentially reducing drug toxicity.
Resistance of cancer cells to chemotherapy can be multifaceted, and understanding the causes could improve the use of existing therapies and potentially reveal new treatment strategies.12 Our recent proteomics studies in childhood acute lymphoblastic leukaemia cells have identified specific protein changes in drug-resistant compared with drug-sensitive cells.13 We identified 10 proteins that differ in structure or quantity between cells that are resistant to vinca alkaloids and those that are sensitive: cytoskeletal proteins (eg, β-tubulin, α-tubulin and actin), proteins that regulate or bind to cytoskeletal proteins (eg, heat shock protein 90β), and proteins involved in RNA processing (eg, heterogeneous nuclear ribonuclear protein-F). Some of these proteins have not previously been associated with drug resistance, and further studies are under way to determine their potential as novel drug targets for treating resistant disease.
In addition, novel antimitotic agents, such as epothilones, are currently undergoing clinical trials in cancer treatment; our studies combined biochemical and proteomic approaches to identify unique changes, such as mutations affecting tubulin, the cellular target of epothilones, in drug-resistant leukaemia cells.14 We found that epothilone-resistant leukaemia cells were cross-resistant to paclitaxel but hypersensitive to vinca alkaloids. This study demonstrates that laboratory studies targeting specific protein alterations can reveal alternative treatment approaches.
Drug resistance is also a major clinical problem in the treatment of many infectious diseases, and, in many cases, the mechanism is unknown. Genetic and protein-sequence data are now available for many microorganisms and are providing tools for understanding their resistance to drugs and for identifying novel agents for treating drug-resistant disease.15,16 For example, azole resistance in Candida albicans has been linked with differential expression of proteins such as Erg10p, a protein involved in the ergosterol biosynthesis pathway.17 This is a potential drug target for the treatment of resistant disease.
Chloroquine has been one of the most successful drugs to treat malaria but has been rendered virtually ineffective in many parts of the world by the widespread emergence of chloroquine resistance. Proteomics technologies are playing a major role in identifying potential therapeutic targets in Plasmodium species, as well as host–pathogen interactions and protein–drug interactions.18 Advances to date include the identification of differences between Plasmodium species, identification of immune targets for vaccination and immune protection, and better understanding of the cellular target(s) of chloroquine and mechanisms of chloroquine resistance.
Proteomics is expected to have a major impact on drug development in the near future. Proteins that are differentially expressed between health and disease are potential drug targets. These can be tested against commercially available libraries of chemical compounds to identify lead compounds — compounds with in vitro activity against the target which are potential new therapies. Exploiting this new knowledge to develop better treatments will be the next challenge.
There are still technical challenges to be overcome. Proteomics must deal with complex mixtures of tissue samples and body fluids. The diversity of the samples is further complicated by the fact that proteins have various isoforms and variants. On average, a single gene can produce up to three proteins. These proteins can then undergo post-translational modifications, such as phosphorylation and glycosylation, which can further increase their functional diversity.
Furthermore, the various gene products and their modifications have differential tissue expression and function. Proteins expressed at low levels in an organism or cell (“low-abundance” proteins, such as transcription factors and some cell-signalling proteins) can often be “swamped” during analysis by “high-abundance” proteins (eg, cytoskeletal proteins, albumin and immunoglobulins). Protein fractionation and enrichment techniques, such as immunodepletion, may concentrate low-abundance proteins for analysis and, combined with the increasing sensitivity of instruments, are starting to overcome this problem.
Proteomic pattern profiling offers enormous hope for early detection of disease, but obstacles remain before it can become commonplace in clinical practice. Mass spectrometry can identify thousands of proteins, and complex algorithms still need to be developed and validated in order to identify specific versus non-specific changes.19 The effects of sample type and quality on proteome pattern and normal variation need to be investigated, while issues of cost and data management need to be resolved. Different samples may have specific storage requirements to preserve protein integrity. Other considerations include the effects of treatment on proteome profiles.
A major challenge is the integration of biochemical, genetic and proteomic data to better understand organisms and disease states. Systems biology is an emerging cross-disciplinary science that aims to integrate the study of organisms in terms of their fundamental structure rather than their individual cellular and molecular make-up. A “system” can be anything from gene regulation in a cell, tissue, organ or organism, to biochemical interactions in a given time and place in a cell. Systems biology requires simultaneous investigation of all interacting components. Proteomics is contributing to this multidisciplinary approach to the study of disease. High-level bioinformatics and computational skills are required to deal with and interpret complex biological systems.
Proteomic analysis of human disease is moving ahead rapidly. Applying its findings will improve our understanding of the roles of individual proteins or entire cellular pathways in the initiation and development of disease.
In 2001, the international Human Proteome Organisation (HUPO) was formed with the vision of consolidating national and regional proteome organisations into a worldwide network, encouraging the spread of proteomics technologies, and disseminating knowledge of the human proteome and proteomes of model organisms. Through the efforts of HUPO and other organisations, databases are being developed of the proteomes of normal and diseased tissues. Initiatives such as these will help accelerate the investigation of disease.
An exciting challenge will be to integrate data from genomic analysis and from the differential, functional and structural studies of proteins and to cross reference them to the biological processes occurring in disease. The development of these databases will expedite high-throughput, cost-effective analysis of clinical samples so that early detection, prognostic, diagnostic and therapeutic tests and strategies will be available to doctors and, ultimately, to patients.
1 Definitions
Genomics The study of the genes of an organism (the “genome”). It includes the systematic use of gene information and the association and regulation of genes to provide answers in biology and medicine. Branches of genomics include genetic analysis, measurement of gene expression, and determination of gene function.
Proteome The entire protein complement of a given cell, tissue or organism.
Proteomics The large-scale study of protein expression, structure and function. The term is analogous to “genomics”, although the proteome is much more complex than the genome. Branches of proteomics include protein separation, identification and quantification, protein sequence analysis, structural proteomics, interaction proteomics and protein modification. In its broader sense, proteomics involves protein activities, modifications, interactions and location in an organism or cell.
2 Development of proteomics
1899 First mass spectrography technique was described by English scientist Thompson.
1970 Two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) was described for separating proteins of Escherichia coli.
1986 Swiss-Prot was established as the first major protein “knowledgebase” it has been maintained collaboratively by European institutions since 1987, including the Department of Medical Biochemistry of the University of Geneva.
1988 Tanaka and colleagues described matrix-assisted laser desorption ionisation (MALDI), which allowed large biomolecules to be analysed more rapidly and efficiently.
1988 Entrez was established as a search and retrieval system for major databases, including protein and genetic databases, by the US National Institutes of Health.
1989 Fenn and colleagues described the application of electrospray ionisation to biomolecules.
1989 Peptide mass fingerprinting (application of mass spectrometry to proteins) was developed as a fast, efficient method to identify frequently observed proteins in electrophoresis gels, but was seldom used because of the need for specialised instruments.
1992 Development of commercial instruments based on MALDI mass spectrometry allowed widespread use of peptide mass fingerprinting.
1999 Quantitative analysis of complex protein mixtures using isotope-coded affinity tags was described.
2001 Highly sensitive protein detection techniques were developed, including difference-gel 2D-electrophoresis (DIGE), a fluorescence-based protein-labelling and separation method; and high throughput screening for serum protein pattern diagnostics using surface enhanced laser desorption/ionisation time of flight (SELDI-TOF) mass spectrometry and protein arrays.
2001 Human Proteome Organisation (www.hupo.org) formed to coordinate and integrate proteome studies around the world.
2002 Nobel Prizes awarded to Fenn (electrospray ionisation) and Tanaka (MALDI).
2003 Immunodepletion techniques were refined and came into routine use; these allow removal of “high-abundance” proteins, giving access to “low-abundance” proteins of interest.
Still to come Development of databases of proteins in different tissues, cells and body fluids in health and disease, and at different disease stages; and databases that link multiple parameters, such as expression of specific proteins and cellular pathways (eg, apoptosis, differentiation, proliferation, migration and invasion), with genetic data and disease states, to assist accurate and rapid diagnosis.
- Maria Kavallaris1
- Glenn M Marshall2
- Children’s Cancer Institute Australia for Medical Research, Sydney, NSW.
We gratefully acknowledge the support of the Children’s Cancer Institute Australia for Medical Research, which is affiliated with the University of New South Wales, and Sydney Children’s Hospital, Sydney, NSW. M Kavallaris is supported by a National Health and Medical Research Council RD Wright Career Development Award.
None identified.
- 1. Fontanarosa PB, DeAngelis CD. Medical applications of biotechnology. JAMA 2005; 293: 866-867.
- 2. Petricoin E, Wulfkuhle J, Espina V, Liotta LA. Clinical proteomics: revolutionizing disease detection and patient tailoring therapy. J Proteome Res 2004; 3: 209-217.
- 3. Ozols RF. Update on the management of ovarian cancer. Cancer J 2002; 8 Suppl 1: S22-S30.
- 4. Petricoin EF, Ardekani AM, Hitt BA, et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet 2002; 359: 572-577.
- 5. Baggerly KA, Morris JS, Edmonson SR, Coombes KR. Signal in noise: evaluating reported reproducibility of serum proteomic tests for ovarian cancer. J Natl Cancer Inst 2005; 97: 307-309.
- 6. Nishizuka S, Chen ST, Gwadry FG, et al. Diagnostic markers that distinguish colon and ovarian adenocarcinomas: identification by genomic, proteomic, and tissue array profiling. Cancer Res 2003; 63: 5243-5250.
- 7. Bahk YY, Kim SA, Kim JS, et al. Antigens secreted from Mycobacterium tuberculosis: identification by proteomics approach and test for diagnostic marker. Proteomics 2004; 4: 3299-3307.
- 8. Ren Y, He QY, Fan J, et al. The use of proteomics in the discovery of serum biomarkers from patients with severe acute respiratory syndrome. Proteomics 2004; 4: 3477-3484.
- 9. Ying W, Hao Y, Zhang Y, et al. Proteomic analysis on structural proteins of severe acute respiratory syndrome coronavirus. Proteomics 2004; 4: 492-504.
- 10. Borozdenkova S, Westbrook JA, Patel V, et al. Use of proteomics to discover novel markers of cardiac allograft rejection. J Proteome Res 2004; 3: 282-288.
- 11. Zhang R, Tremblay TL, McDermid A, et al. Identification of differentially expressed proteins in human glioblastoma cell lines and tumors. Glia 2003; 42: 194-208.
- 12. Verrills NM, Kavallaris M. Drug resistance mechanisms in cancer cells: a proteomics perspective. Curr Opin Mol Ther 2003; 5: 258-265.
- 13. Verrills NM, Walsh BJ, Cobon GS, et al. Proteome analysis of vinca alkaloid response and resistance in acute lymphoblastic leukemia reveals novel cytoskeletal alterations. J Biol Chem 2003; 278: 45082-45093.
- 14. Verrills NM, Flemming CL, Liu M, et al. Microtubule alterations and mutations induced by desoxyepothilone B: implications for drug-target interactions. Chem Biol 2003; 10: 597-607.
- 15. Haney SA, Alksne LE, Dunman PM, et al. Genomics in anti-infective drug discovery — getting to endgame. Curr Pharm Des 2002; 8: 1099-1118.
- 16. Schmidt FR. The challenge of multidrug resistance: actual strategies in the development of novel antibacterials. Appl Microbiol Biotechnol 2004; 63: 335-343.
- 17. Hooshdaran MZ, Barker KS, Hilliard GM, et al. Proteomic analysis of azole resistance in Candida albicans clinical isolates. Antimicrob Agents Chemother 2004; 48: 2733-2735.
- 18. Johnson JR, Florens L, Carucci DJ, Yates JR. Proteomics in malaria. J Proteome Res 2004; 3: 296-306.
- 19. Johann DJ, McGuigan MD, Patel AR, et al. Clinical proteomics and biomarker discovery. Ann N Y Acad Sci 2004; 1022: 295-305.
Abstract
Since the human genome was sequenced, there has been intense activity to understand the function of the 30 000 identified genes; attention has now turned to the products of genes — proteins.
Proteomics is the large-scale study of the structure and function of proteins; it includes the rapidly evolving field of disease proteomics, which aims to identify proteins involved in human disease and to understand how their expression, structure and function cause illness.
Proteomics has identified proteins that offer promise as diagnostic or prognostic markers, or as therapeutic targets in a range of illnesses, including cancer, immune rejection after transplantation, and infectious diseases such as tuberculosis and malaria; it has the potential to allow patient-tailored therapy.
Some major challenges remain, both technical (eg, detecting “low-abundance” proteins, and maintaining sample stability) and in data management (eg, correlating changes in proteins with disease processes).