




We report a young woman presenting with digital gangrene, paranasal sinusitis, mononeuritis multiplex, and rapidly progressive glomerulonephritis without asthma and eosinophilia — an extremely rare variant of this disease.
Churg–Strauss syndrome (CSS), or allergic granulomatosis and angiitis, is a granulomatous vasculitis affecting small- to medium-sized arteries and veins. Although CSS is a systemic disease characterised by asthma and eosinophilia, various limited forms have been described.1 Here, we report a patient with such a limited form of the disease.
A 35-year-old woman presented with a 15-day history of gangrene of the fingertips of her left hand, and weakness of the right wrist joint and the left ankle joint. She had a 3-year history of recurrent paranasal sinusitis. There was no history of asthma or any other allergic disorder, parasitic infestation, or smoking.
On presentation, the patient had a pulse rate of 78 beats/min, blood pressure of 150/90 mm/Hg, and a temperature of 38.8°C. She had conjunctival pallor, gangrene involving all fingertips of the left hand, palpable purpura on the anterior aspect of the left leg, and 3/5 grade motor weakness of the right wrist joint and left ankle joint on dorsiflexion. The provisional diagnosis was systemic vasculitis syndrome. Blood tests on presentation showed (reference ranges in parentheses) a haemoglobin level of 93 g/L (110–165 g/L), a white blood cell count of 15 × 109 cells/L (4–11 × 109 cells/L), an eosinophil count of 0.15 × 109 cells/L (0–0.4 × 109 cells/L), and erythrocyte sedimentation rate of 35 mm/h (< 15 mm/h). Levels of urea, creatinine and electrolytes, fibrinogen level and prothrombin time, and the results of liver function tests, were within normal ranges. Urinalysis showed 3+ proteinuria (3 g/L) and microscopic analysis showed 30–40 red blood cells per high power field (reference range, < 4). The creatinine clearance rate was 98 mL/min.
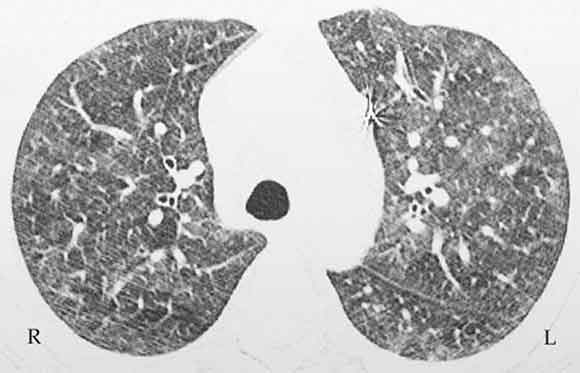
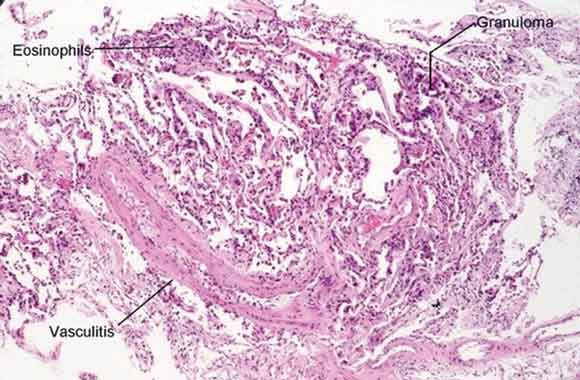
A chest x-ray demonstrated bilateral ground glass infiltrates, predominantly in the basal zones. High-resolution computed tomography (CT) of the chest revealed bilateral patchy ground glass opacities (Box 1). Spirometry showed a forced vital capacity (FVC) of 65% predicted, a forced expiratory volume (FEV1) of 68% predicted, and FEV1/FVC of 104%. An echocardiogram was normal. A nerve conduction velocity study showed axonal involvement, with reduced nerve conduction velocity in the right radial and left superficial peroneal nerves. CT of the paranasal sinuses showed chronic pansinusitis. Histopathological examination of the open-lung biopsy showed leukocytoclastic vasculitis, granuloma formation, and extravascular eosinophilic infiltration (Box 2). Renal biopsy showed pauci-immune, crescentic glomerulonephritis. Renal vessel Doppler ultrasonography and renal angiography gave normal results and ruled out any renal vessel involvement. The patient tested positive for perinuclear antineutrophilic cytoplasmic antibodies (ANCAs) and negative for cytoplasmic ANCAs by the indirect immunofluorescence method. A more specific enzyme-linked immunosorbent assay (ELISA) for anti-myeloperoxidase was positive, and another, for anti-proteinase 3, was negative. These tests ruled out Wegner’s granulomatosis. Antinuclear factor, anticardiolipin antibodies, hepatitis B surface antigen, and serological tests for hepatitis C and HIV were negative, thus ruling out significant secondary causes of vasculitis.
The patient was treated with prednisolone (1 mg/kg per day, orally) and her condition showed marked clinical as well as radiological improvement. She became afebrile after 4 days, normotensive after 4 weeks, and her palpable purpura disappeared after 6 weeks. High-resolution CT of the chest, repeated at 4 and 8 weeks, showed progressive resolution of ground glass infiltrates. Repeat urinalysis showed resolution of proteinuria and haematuria at 8 weeks. The prednisolone dose was tapered over 8 weeks to a maintenance dose of 0.125 mg/kg every second day. Additional management included surgical amputation of all left-hand fingertips and physiotherapy for the motor weakness.
Our patient presented without the asthma and eosinophilia which are hallmark features of CSS. To the best of our knowledge, this is only the second report of this disease presenting in this way. A previous case report from Turkey described a young patient with CSS but no asthma and eosinophilia.2 CSS with eosinophilia but without asthma is also extremely rare and few cases have been described.3-7
In 1990, the American College of Rheumatology proposed six criteria for classification of CSS. These include asthma, eosinophilia (more than 10%), paranasal sinusitis, pulmonary infiltrates on chest x-ray (may be transient), extravascular eosinophils on biopsy, and mononeuritis multiplex or polyneuropathy.8 A patient is said to have CSS if at least four of these six criteria are met. The presence of any four or more of these criteria yields a sensitivity of 85% and a specificity of 99.7%. Our patient had four of the six features (paranasal sinusitis, mononeuritis multiplex, extravascular eosinophilic infiltration on biopsy, and pulmonary infiltrates on chest radiography).
As illustrated by this case, CSS may present without asthma and eosinophilia. Earlier recognition of this disease and treatment with corticosteroids and/or immunosuppressive agents is important to prevent life-threatening visceral involvement.
- 1. Lie JT. Limited forms of Churg-Strauss syndrome. Pathol Annu 1993; 28: 199-220.
- 2. Sevinc A, Hasanoglu HC, Gokirmak M, et al. Allergic granulomatosis and angiitis in the absence of asthma and blood eosinophilia: a rare presentation of limited Churg-Strauss syndrome. Rheumatol Int 2004; 24: 301-304.
- 3. Malik TQ, Youmbissi TJ, Gacha R, et al. Atypical presentation of Churg-Strauss syndrome: another “forme fruste” of the disease? Am J Med Sci 2002; 324: 276-278.
- 4. Morimatsu Y, Kinoshita M, Koga T, et al. A case of allergic granulomatosis and angiitis without symptoms of asthma. Nihon Kokyuki Gakkai Zasshi 2003; 41: 655-659.
- 5. Jessurun J, Azevedo M, Saldana M. Allergic angiitis and granulomatosis (Churg-Strauss syndrome): report of a case with massive thymic involvement in a nonasthmatic patient. Hum Pathol 1986; 17: 637-639.
- 6. Chen KR, Ohata Y, Sakurai M, et al. Churg-Strauss syndrome: report of a case without preexisting asthma. J Dermatol 1992; 19: 40-47.
- 7. Yamashita Y, Yorioka N, Taniguchi Y, et al. Nonasthmatic case of Churg-Strauss syndrome with rapidly progressive glomerulonephritis. Intern Med 1998; 37: 561-563.
- 8. Masi AT, Hunder GG, Lie JT, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 1990; 33: 1094-1100.
None identified.