





Volume 181 - Issue 6
Clinical usefulness of plasma homocysteine in vascular disease
Authors: Graeme J Hankey, John W Eikelboom, Wai Khoon Ho and Frank M van Bockxmeer
Med J Aust 2004; 181 (6): 314-318. || doi: 10.5694/j.1326-5377.2004.tb06296.x
Published online: 20 September 2004
Published online: 20 September 2004
Abstract
Raised plasma homocysteine (tHcy) concentrations are caused by genetic mutations, vitamin deficiencies, renal and other diseases, numerous drugs, and increasing age.
Raised tHcy concentrations are associated with laboratory evidence of atherogenesis (eg, endothelial dysfunction) and thrombosis, and epidemiological evidence of an increased risk of atherothrombotic vascular disease.
An association between raised tHcy concentration and an increased risk of atherothrombosis is independent of other vascular risk factors, strong, dose-related and biologically plausible, but has not been proven to be causal in randomised controlled trials.
A recent trial identified no significant benefit from lowering tHcy concentration by folic-acid-based multivitamin therapy among 3680 patients with recent ischaemic stroke, but did not reliably exclude a modest but important reduction in the relative risk of stroke of up to 20%; a difference of only 2 mmol/L in tHcy concentration between the two treatment groups was probably due to widespread vitamin use and fortification of grains and staple foods with folate in North America.
There is currently insufficient evidence to recommend routine screening and treatment of high tHcy concentrations with folic acid and other vitamins to prevent atherothrombotic vascular disease.
Homocysteine is a sulfhydryl-containing amino acid derived from the essential amino acid methionine, which is abundant in animal sources of protein.1 The metabolic pathway that converts methionine to homocysteine (Box 1) is essential for the proper functioning of many biomolecules, including DNA, proteins, phospholipids and neurotransmitters.1-3
Plasma concentrations of homocysteine vary widely, but intracellular concentrations of homocysteine are normally maintained within a relatively narrow range.3 Total plasma (or total serum) homocysteine (tHcy) refers to the combined pool of free, bound, reduced and oxidised forms of homocysteine in the blood.1
Factors that influence homocysteine metabolism and cause hyperhomocysteinaemia
Causes of hyperhomocysteinaemia are multifactorial (Box 2).4,5 Most operate by altering the function or blood concentrations of B vitamins (folic acid, vitamin B12, vitamin B6) involved as cofactors in the homocysteine metabolic pathway, interfering with renal function, or influencing enzyme activities. The single most important determinant of tHcy in the general population is folate status.3
Lowering plasma homocysteine concentrations by
folic-acid-based vitamin supplementation
The Homocysteine Lowering Trialists’ Collaboration meta-analysis of 12 clinical trials, including 1114 individuals without renal failure, most of whom had normal folate status, showed that between 0.5 mg and 5 mg of folic acid daily lowers tHcy levels by 25% (95% CI, 23%–28%).6 The minimum effective daily dose of folic acid for achieving maximal homocysteine-lowering efficacy is about 0.5 mg. Vitamin B12 (0.02–1 mg daily) further reduces tHcy levels by about 7% (95% CI, 3%–10%). Vitamin B6 (2–50 mg daily) and betaine (a methyl-group donor involved in the metabolism of methionine) are less effective.7
Measurement of plasma homocysteine
The most robust measure of tHcy level is a fasting tHcy test. Testing with methionine loading may be more sensitive for detecting mild disturbances in homocysteine metabolism, but its clinical usefulness is uncertain, and a large dose of methionine may very rarely cause a potentially lethal metabolic/toxic encephalopathy.8
Specimen collection and processing
Blood specimens are usually collected in EDTA (ethylenediaminetetraacetic acid) tubes, but collection in heparinised tubes will not materially influence the results. Compared with optimally prepared plasma, tHcy levels obtained with citrated plasma are 5%–15% lower, and in serum are 5%–15% higher.4
Blood specimens should be centrifuged within 1 hour or kept on ice until centrifugation within 8 hours.4 Uncentrifuged blood that has been kept at room temperature for more than two hours is of little value.4
Measurement
Chromatographic assays may be best suited for laboratories with experience in this technique, whereas enzyme assays and immunoassays are likely to be suitable for any laboratory.4
Definition of hyperhomocysteinaemia
An elevated plasma tHcy level (hyperhomocysteinaemia) is most commonly defined according to arbitrary cut-off points (eg, 95th percentile) in the distribution of values obtained from the so-called normal population. Each laboratory should establish reference limits for its own region, with separate reference limits for children, adults, the elderly, and pregnant women (Box 3). For populations taking folic acid supplements or eating a folic-acid-fortified diet, the upper reference limit is generally 20%–25% lower than for populations not receiving extra folic acid.4
Homocysteine and atherothrombosis
The concept that hyperhomocysteinaemia may predispose to atherothrombosis evolved more than 30 years ago, when patients with homocystinuria (and extremely high plasma concentrations of tHcy) were observed to have a high incidence of vascular disease and premature death from myocardial infarction.10 This has since provoked research to determine whether hyperhomocysteinaemia is a causal risk factor for atherothrombosis.
Laboratory evidence
Laboratory studies suggest that an elevated tHcy level is both atherogenic and thrombogenic.1,11-13 The proposed mechanisms by which hyperhomocysteinaemia produces complex changes in the structure and function of cerebral, coronary and peripheral vessels are listed in Box 4.
Epidemiological evidence
Observational studies: A recent meta-analysis by the Homocysteine Studies Collaboration of individual patient data from 12 prospective and 18 retrospective observational studies of 16 786 individuals in healthy populations found that, after adjusting for confounding caused by known vascular risk factors and correction for regression dilution caused by random variation in homocysteine measurements, a 25% higher than usual concentration of Hcy in the blood (about 3 μmol/L [0.41 mg/L]) was associated with 11% greater odds of ischaemic heart disease (odds ratio [OR], 0.89; 95% CI, 0.83–0.96), and 19% greater odds of stroke (OR, 0.81; 95% CI, 0.69–0.95).14
An independent and concurrent meta-analysis of 20 prospective studies of serum homocysteine and risk of vascular disease involving 3820 participants found that a 5 μmol/L increase in tHcy level was associated with a 32% increase in odds of ischaemic heart disease (OR, 1.32; 95% CI, 1.19–1.45) and a 59% increase in odds of stroke (OR, 1.59; 95% CI, 1.29–1.96).15
There were some inconsistencies in the results obtained by different study methods. Stronger associations between tHcy level and vascular risk were found in studies that used less robust methods, and smaller associations or no association were reported by more robust prospective cohort studies. In addition, the temporal relationship between the onset of elevated tHcy level and atherothrombotic vascular events is unclear; the finding of a stronger association in case–control studies compared with cohort studies suggests that elevated tHcy level may rise after an acute vascular event in response to tissue damage or tissue repair.16
Genetic studies: A meta-analysis of 40 observational studies involving 11 162 people with coronary heart disease and 12 758 control subjects showed that individuals with the MTHFR 677 TT genotype (MTHFR 677 C →T polymorphism of the gene that encodes the MTHFR enzyme), who have 20% higher tHcy levels than normal, have a 16% (95% CI, 5%–28%) greater risk of ischaemic heart disease than those with the CC polymorphism.17
An independent meta-analysis of 72 studies in which the prevalence of a mutation in the MTHFR gene was determined in 16 849 people showed that a 5 mmol/L increase in tHcy level was associated with a 42% increase in odds of ischaemic heart disease (OR, 1.42; 95% CI, 1.11–1.84), a 65% increase in odds of stroke (OR, 1.65; 95%CI, 0.66–4.13), and a 60% increase in odds of venous thromboembolism (OR, 1.60; 95% CI, 1.15–2.22).15
Dietary studies: Large observational studies correlating diet with long-term risk of vascular events among more than 50 000 healthy individuals suggest that a decreased dietary intake of folate is associated with an increased risk of ischaemic stroke and cardiovascular events, independent of major lifestyle and other dietary factors.18,19
Clinical trial evidence
To date, several randomised controlled trials have investigated the effect of folic acid and/or vitamins B6 and B12 on multiple surrogate markers of cardiovascular disease, and one has evaluated the effect on clinical outcome events, such as stroke, myocardial infarction, or death.
Endothelial function: Folic acid has been shown to prevent postprandial endothelial dysfunction in normohomocysteinaemic subjects, and to improve endothelial function in patients with hyperhomocysteinaemia, hypercholesterolaemia, diabetes and coronary artery disease.3 The exact mechanisms underlying the ameliorative effects of folate on the endothelium are uncertain, but may include homocysteine-lowering, antioxidant actions, effects on cofactor availability, or direct interactions with the enzyme endothelial nitric oxide synthase.11-13
Carotid artery intimal thickness: Two studies have reported a decrease in the rate of progression of carotid artery intimal thickness in patients taking oral folic-acid-based multivitamins compared with before supplementation.20,21 In addition, a double-blind randomised trial of 56 stable renal transplant recipients with documented hyperhomocysteinaemia who were randomly assigned to vitamin supplementation with folic acid (5 mg daily), vitamin B6 (50 mg daily) or vitamin B12 (400 μg daily) or placebo for 6 months showed a significant decrease in carotid intima media thickness in patients who received vitamins (0.95 ± 0.20 mm [before] v 0.64 ± 0.17 mm [after]; P < 0.0001), but a significant increase in those who received placebo (0.71 ± 0.16 mm [before] v 0.87 ± 0.19 mm [after]; P < 0.05).22
Exercise electrocardiography: A double-blind, placebo-controlled, randomised trial of 158 healthy siblings of patients with premature clinical atherothrombotic disease and raised tHcy concentrations who were randomly assigned placebo or folic acid (5 mg daily) and vitamin B6 (250 mg daily) for 2 years showed that vitamin treatment was associated with a decrease in fasting tHcy level (14.7 mmol/L [placebo] v 7.4 mmol/L [vitamins]) and a decreased rate of abnormal exercise electrocardiography test results (OR, 0.40; 95% CI, 0.17–0.93; P = 0.035).23
Coronary artery restenosis and revascularisation: Following coronary angioplasty, 205 patients were allocated to placebo or the combination of folic acid (1 mg), vitamin B12 (0.4 mg), and pyridoxine (10 mg) in a randomised, double-blind trial.24 After 6 months, multivitamin treatment was associated with a significant reduction in plasma tHcy level (11.1 mmol/L [placebo] v 7.2 mmol/L [vitamins], P < 0.001) and the rate of coronary restenosis (37.6% [placebo] v 19.6% [vitamins]; P = 0.01),25 a benefit that remained evident at 1 year.26
However, the most recent study randomly allocated 626 patients undergoing coronary stenting to a single intravenous dose followed by daily oral placebo or the combination of folic acid (1.2 mg), vitamin B12 (0.06 mg), and pyridoxine (48 mg), and after 6 months patients assigned multivitamin treatment had significantly smaller minimum mean coronary artery lumen diameters, the primary end point (1.74 mm [placebo] v 1.59 mm [vitamins]), and higher rates of restenosis (27% v 35%).26
Serious clinical vascular events: The Vitamins in Stroke Prevention (VISP) Trial randomly allocated 3680 North American and Scottish patients with recent (3–120 days) non-disabling, non-cardiogenic, ischaemic stroke, who had baseline blood concentrations of homocysteine above the 25th percentile of normal, to double-blind treatment with a high-dose multivitamin formulation (including 2.5 mg folic acid, 0.4 mg cobalamin and 25 mg pyridoxine) or a low-dose formulation (including 20 μg folic acid, 6 mg cobalamin and 200 mg pyridoxine).27 After 2 years of follow-up, plasma tHcy concentration was 2 mmol/L lower in the high-dose group, but there was no significant difference in the cumulative incidence of the primary outcome event, recurrent cerebral infarction (8.8% [low dose] v 9.2% [high dose]; relative risk [RR], 1.0; 95% CI, 0.8–1.3; P = 0.68). There was also no significant reduction in any coronary event (7.4% v 7.0%; RR, 0.9; 95% CI, 0.7–1.2), death (6.9% v 5.9%; RR, 0.9; 95% CI, 0.7–1.1), and the combined outcome of ischaemic stroke, coronary heart disease or death (18.6 v 18.0; RR, 0.9; 95% CI, 0.7–1.1).27
The (unexpectedly) small between-group difference of 2 mmol/L in plasma tHcy concentration is likely to reflect the high prevalence of vitamin supplement use and widespread fortification of the grain supply and staple foods in North America with folate since the inception of the VISP trial.28 Grain and food fortification is likely to have reduced the statistical power of VISP substantially.29 Furthermore, the vitamin regimen used in VISP may have contained too little vitamin B12 in the high-dose group for elderly patients with poor B12 absorption, and too much in the low-dose group (more than the recommended daily intake), because, in the presence of folate repletion, blood concentrations of tHcy are highly dependent on vitamin B12.30 The lower than anticipated rates of recurrent strokes in both study groups and short follow-up period (only 2 years) also limited the statistical power of the VISP trial to reliably identify or exclude a modest, but clinically important, therapeutic effect of vitamins.
Interpretation of the evidence
The features in favour of a causal relationship between tHcy concentration and atherothrombosis are that the prospective observational and genetic studies did not share the same potential sources of error, but all yielded highly significant results consistent with a strong, dose-related, independent, and biologically plausible association between increasing tHcy concentration (resulting from impaired folate metabolism) and increasing risk of atherothrombotic vascular disease. Moreover, randomised controlled trials indicate that reducing plasma Hcy concentration by means of multivitamin therapy produces favourable effects on multiple surrogate markers of vascular disease. These data suggest that, if the association is causal, lowering tHcy concentration by 3 mmol/L from current concentrations would be expected to reduce the risk of ischaemic heart disease by 16% (95% CI, 11%–20%), stroke by 24% (95% CI, 15%–33%) and venous thromboembolism by 25% (95% CI, 8%–38%).15
However, there are several features casting doubt on a causal relationship.
Inconsistency in the results of epidemiological studies obtained by different methods (with smaller associations or no association in more robust studies).
Uncertainty about the temporal relationship between the onset of elevated tHcy concentration and the onset of vascular events (eg, elevated tHcy concentration may be a marker of acute tissue ischaemia16).
Uncertainty as to whether raised tHcy concentration is a marker of another risk factor for atherothrombosis (eg, renal impairment, smoking, alcohol, metabolic syndrome, folate deficiency, and defective methylation because of altered ratios of S-adenosylmethionine [a direct methyl-group donor] and S-adenosylhomocysteine [a potent inhibitor of transmethylation reactions]).
Lack of reliable evidence from randomised controlled trials that lowering plasma tHcy concentration prevents “hard” vascular outcome events. The VISP trial was the first large RCT evaluating the effect of folic-acid-based multivitamin therapy on “hard” clinical outcomes to be completed.27 Although the estimate of the treatment effect on recurrent cerebral infarction was “null” (RR, 1.0; 95% CI, 0.8–1.3), the estimate is imprecise, and its 95% confidence interval is consistent with up to a 20% reduction in RR, and a 30% reduction in the RR of death, associated with lowering tHcy concentration by 2 mmol/L.27 Greater reductions in tHcy concentration may be associated with even greater reduction in risk of major vascular events.
Implications for research
There are at least 12 large ongoing clinical trials involving a combined total of more than 50 000 patients which are testing the homocysteine hypothesis. These trials need to be completed, and individual patient data submitted for meta-analysis by the Homocysteine Lowering Trialists’ Collaboration (Robert Clarke, Clinical Trials Service Unit, Oxford, UK <www.ctsu.ox.ac.uk>) to more reliably and precisely estimate the effect of homocysteine-lowering treatment on the risk of atherothrombotic vascular disease, as well as its safety.
Implications for clinicians
Folic-acid-based multivitamin therapy has been proven neither effective nor ineffective in preventing clinical vascular events — there are simply not enough reliable data from large randomised controlled trials.
Vitamin therapy is costly and potentially risky. Repeated doses exceeding 400 mg daily of vitamin B6 may cause a sensory peripheral neuropathy, and folic acid therapy may cause progressive neurological damage (subacute combined degeneration of the spinal cord) in individuals with subclinical vitamin B12 deficiency. Although the latter can be avoided by excluding B12 deficiency before starting folic acid therapy, or supplementing folic acid therapy with at least 400 mg per day of vitamin B12, it is important to not assume that folic-acid-based multivitamin therapy is safe for everyone.
The antioxidant nutrient beta-carotene was assumed to be safe and effective in preventing atherothrombotic vascular disease until two trials in heavy smokers showed that it was associated with a higher incidence of lung cancer and higher all-cause mortality.31
While awaiting the results and meta-analysis of the ongoing clinical trials of folic-acid-based homocysteine-lowering multivitamin therapy, insufficient evidence exists to recommend routine screening and treatment of high tHcy concentration with folic acid and other vitamins to prevent atherothrombotic vascular disease.
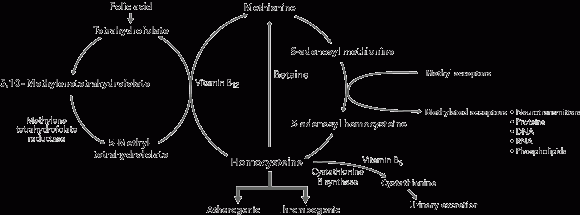
1 Homocysteine metabolic pathways
In the methylation pathway, homocysteine acquires a methyl group either from betaine (a reaction that occurs mainly in the liver) or from 5-methyltetrahydrofolate (a reaction that occurs in all tissues and is vitamin B12-dependent). In the transsulfuration pathway, homocysteine is metabolised to cystathionine in a reaction catalysed by cystathionine β synthase and requiring vitamin B6.
2 Causes of hyperhomocysteinaemia
Genetic factors
5,10-Methylenetetrahydrofolate reductase C677T homozygosity (common)
Heterozygosity for cystathionine β synthase defects (uncommon)
Homocystinuria (very rare)
Physiological factors
Increasing age
Male sex
Menopause
Reduced glomerular filtration rate
Increased muscle mass
Lifestyle factors
Reduced vitamin intake (folate, vitamin B12, vitamin B6)
Smoking
Coffee
Alcohol consumption
Physical inactivity
Disease states
Vitamin deficiency (folate, vitamin B12, vitamin B6)
Renal failure
Hypothyroidism
Diabetes mellitus
Psoriasis
Malignancies
Drugs
Lipid lowering* — cholestyramine, nicotinic acid, fibric acid derivatives (eg, fenofibrate)
Anticonvulsants — phenytoin, carbamazepine
Sex hormones — androgens
Anti-rheumatic drugs — methotrexate
Other — cyclosporin, diuretics, levodopa, methionine loading (oral, intravenous, peritoneal), theophylline, trimethoprim
* HMG-CoA reductase inhibitors have no effect on total plasma homocysteine levels.
3 Upper limit of reference ranges for fasting total plasma (or total serum) homocysteine level*
Population |
Folate supplemented |
Not folate supplemented |
|||||||||||||
Children (< 15 years) |
8 μmol/L |
10 μmol/L |
|||||||||||||
Adults (15–65 years) |
12 μmol/L |
15 μmol/L |
|||||||||||||
Elderly (> 65 years) |
16 μmol/L |
20 μmol/L |
|||||||||||||
Pregnant women |
8 μmol/L |
10 μmol/L |
|||||||||||||
* Reproduced with permission from Moat et al3 |
|||||||||||||||
4 In vitro effects of homocysteine that may be relevant to atherogenesis and thrombogenesis
Atherogenesis
Induces DNA hypomethylation and expression of genes known to mediate cell growth and differentiation
Induces oxidative stress
Induces vascular inflammation by altering expression of tumour necrosis factor-α and inducible NO synthase
Induces endothelial dysfunction as a result of increased oxidative stress, increased ADMA, decreased bioavailability of nitric oxide (due to increased oxidative stress), and increased inflammation
Alters hepatic and macrophage lipoprotein metabolism, in part by enhancing uptake of modified low density lipoprotein
Induces hypertrophy and altered mechanics in the microcirculation, and increases intima media thickness
Thrombogenesis
Induces tissue factor expression in monocytes
Modulates leukocyte–endothelium interactions
Increases platelet aggregation
Enhances binding of lipoprotein (a) to fibrin
Interferes with several clotting factors
Competing interests
None identified.
References
- Hankey GJ, Eikelboom JW. Homocysteine and vascular disease. Lancet 1999; 354: 407-413. i1085853
- D’Angelo A, Selhub J. Homocysteine and thrombotic disease. Blood 1997; 90: 1-11.
- Moat SJ, Lang D, McDowell IFW, et al. Folate, homocysteine, endothelial function and cardiovascular disease. J Nutr Biochem 2004; 15: 64-79. i1085856
- Refsum H, Smith AD, Ueland PM, et al. Facts and recommendations about total homocysteine determinations: an expert opinion. Clin Chem 2004; 50: 3-32. i1085858
- Desouza C, Keebler M, McNamara DB, Fonseca V. Drugs affecting homocysteine metabolism. Impact on cardiovascular risk. Drugs 2002; 62: 605-616. i1085860
- Homocysteine lowering trialist’s collaboration. Lowering blood homocysteine with folic acid based supplements: meta-analysis of randomised trials. BMJ 1998; 316: 894-898. i1085862
- Van Guldener C, Janssen MJ, deMeer K, et al. Effect of folic acid and betaine on fasting and methionine-loading plasma homocysteine and methionine concentration in chronic haemodialysis patients. J Intern Med 1999; 245: 175-183. i1085864
- Cottington EM, LaMantia C, Stabler SP, et al. Adverse event associated with methionine loading test. A case report. Arterioscler Thromb Vasc Biol 2002; 22: 1046-1050. i1085866
- Ubbink JB. Assay methods for the measurement of total homocyst(e)ine in plasma. Semin Thromb Hemost 2000; 26: 233-241. i1085868
- McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol 1969; 56: 111-128. i1085870
- Symons JD, Mullick AE, Ensunsa JL, et al. Hyperhomocysteinaemia evoked by folate depletion: effects on coronary and carotid arterial function. Arterioscler Thromb Vasc Biol 2002; 22: 772-780. i1085872
- Faraci FM. Hyperhomocysteinaemia: a million ways to lose control. Arterioscler Thromb Vasc Biol 2003; 23: 371-373.
- Faraci FM, Lentz SR. Hyperhomocysteinemia, oxidative stress, and cerebral vascular dysfunction. Stroke 2004; 35: 345-347. i1085875
- The Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke. A meta-analysis. JAMA 2002; 288: 2015-2022. i1085877
- Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ 2002; 325: 1202-1206. i1085879
- Dudman NP: An alternative view of homocysteine. Lancet 1999; 354: 2072-2074. i1085881
- Klerk M, Verhoef P, Clarke R, et al, and the MTHFR Studies Collaboration Group. MTHFR 677CÆT polymorphism and risk of coronary heart disease. A meta-analysis. JAMA 2002; 288: 2023-2031. i1085883
- Bazzano LA, He J, Ogden LG, et al. Dietary intake of folate and risk of stroke in US men and women. NHAMES I Epidemiologic follow-up study. Stroke 2002; 33: 1183-1189. i1085885
- He K, Merchant A, Rimm EB, et al. Folate, vitamin B6, and B12 intakes in relation to risk of stroke among men. Stroke 2004; 35: 169-174. i1085887
- Peterson JC, Spence JD. Vitamins and progression of atherosclerosis in hyper-homocyst(e)inaemia [letter]. Lancet 1998; 351: 263. i1085889
- Hackam DG, Peterson JC, Spence JD. What level of plasma homocyst(e)ine should be treated? Effects of vitamin therapy on progression of carotid atherosclerosis in patients with homocyst(e)ine levels above and below 14 μmol/L. Am J Hypertens 2000; 13: 105-110. i1085891
- Marcuci R, Zanazzi M, Bertoni E, et al. Vitamin supplementation reduces the progression of atherosclerosis in hyperhomocysteinaemic renal-transplant recipients. Transplantation 2003; 75: 1551-1555. i1085893
- Vermeulen EGJ, Stehouwer CDA, Twisk JWR, et al. Effect of homocysteine-lowering treatment with folic acid plus vitamin B6 on progression of subclinical atherosclerosis: a randomised, placebo-controlled trial. Lancet 2000; 355: 517-522. i1085895
- Schnyder G, Roffi M, Pin R, et al. Decreased rate of coronary restenosis after lowering of plasma homocysteine levels. N Engl J Med 2001; 345: 1593-1600. i1085897
- Schnyder G, Roffi M, Flammer Y, et al. Effect of homocysteine-lowering therapy with folic acid, vitamin B12, and vitamin B6 on clinical outcome after percutaneous coronary intervention. JAMA 2002; 288: 973-979. i1085899
- Lange H, Suryapranata H, De luca G, et al. Folate therapy and in-stent restenosis after coronary stenting. New Engl J Med 2004; 350: 2673-2681. i1085901
- Toole JF, Malinow R, Chambless L, et al. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction and death. The Vitamin Intervention for Stroke Prevention (VISP) randomised controlled trial. JAMA 2004; 291: 565-575. i1085903
- Jacques PF, Selhub J, Bostom AG, Wilson PW, et al. The effect of folic acid fortification on plasma folate and total homocysteine concentrations. N Engl J Med 1999; 340: 1449-1454. i1085905
- Bostom AG, Selhub J, Jacques PF, Rosenberg IH. Power shortage: clinical trials testing the “homocysteine hypothesis” against a background of folic acid-fortified cereal grain flour. Ann Intern Med 2001; 135: 133-137. i1085907
- Quinlivan EP, McPartlin J, McNulty H, et al. Importance of both folic acid and vitamin B12 in reduction of risk of vascular disease. Lancet 2002; 359: 227-228. i1085909
- Morris CD, Carson S. Routine vitamin supplementation to prevent cardiovascular disease: a summary of the evidence for the US Preventive Services Task Force. Ann Intern Med 2003; 139: 56-70. i1085911