





Volume 181 - Issue 5
Paget’s disease of bone
Author: John P Walsh
Med J Aust 2004; 181 (5): 262-265. || doi: 10.5694/j.1326-5377.2004.tb06265.x
Published online: 6 September 2004
Published online: 6 September 2004
Abstract
Paget’s disease of bone is common, affecting up to 4% of Australians over the age of 55 years. The incidence of the disease and the severity of newly diagnosed cases appear to be falling, for unknown reasons.
The cause of Paget’s disease is unknown, but there is a strong genetic influence. Recently, mutations in the sequestosome 1/p62 gene have been identified as a cause of familial Paget’s disease and of some apparently sporadic cases of the disease.
The disease is often asymptomatic, but can cause bone pain, deformity, fracture and other complications.
Paget’s disease is eminently treatable. Potent bisphosphonates such as pamidronate, alendronate and risedronate relieve symptoms and may reduce the risk of complications.
The Pharmaceutical Benefits Scheme subsidises treatment only for patients with symptomatic disease. A strong case be made for also treating asymptomatic patients with involvement of long bones, vertebrae or base of skull, patients with significant osteolytic lesions, and perhaps all younger patients.
Paget’s disease of bone is a chronic disorder, characterised by focal areas of excessive osteoclastic bone resorption accompanied by a secondary increase in osteoblastic bone formation. The classic description of the disease was by Sir James Paget in 1877 in a paper describing the clinical features of a disorder he called osteitis deformans.1 The disease results in bone expansion and structural weakness, which can cause pain, deformity, and a range of complications. Here, I review recent advances in understanding of the disease, as well as practical management. For more detailed reviews, see two recently published sets of consensus management guidelines.2,3
Epidemiology and pathogenesis of Paget’s disease
Paget’s disease predominantly affects the elderly. It is uncommon in people under the age of 55 years and is more common in males than females, with a sex ratio of 1.8 : 1. There are marked geographical differences in prevalence. The disease is most common in the United Kingdom (particularly Lancashire), followed by Australia, New Zealand and North America, in part reflecting the large number of British migrants to those countries.4 By contrast, the disease is uncommon in Scandinavia, Africa and Asia. A radiographic study carried out in Western Australia in 1978 reported a prevalence of 4% in people aged over 55 years.5 Recent studies from the United Kingdom and New Zealand suggest that the incidence of Paget’s disease is falling, perhaps by as much as 50% over the past 20 years, and that the severity of newly diagnosed cases is falling.6,7 The reason for this is unknown. There are no recent prevalence studies from Australia.
The cause of Paget’s disease is unknown. There is a strong genetic component, and 15%–20% of those affected have a first-degree relative with the disease.8,9 Genome linkage scans have identified several loci associated with familial Paget’s disease. Recently, mutations in the sequestosome 1/p62 gene have been identified in about a third of affected kindreds, and in 5%–15% of patients with no family history of the disorder.10-12 The SQSTM1/p62 protein is a selective activator of the transcription factor NFB, which plays an important role in osteoclast differentiation and activation in response to the cytokines RANK-ligand and interleukin-1.13,14 Mutations in the SQSTM1/p62 gene are therefore a plausible cause of Paget’s disease, but it is unclear how germline DNA mutations (present in every osteoclast) cause bone disease that is focal in nature.
Environmental factors may also play a role. Several studies have postulated that viruses, particularly paramyxoviruses such as canine distemper or measles virus, play a role in pathogenesis, but definitive evidence for this is lacking.15 Presumably, the declining incidence of Paget’s disease reflects a decline in one or more as yet unidentified environmental influences.
Diagnosis
Clinical features
Paget’s disease presents clinically in a variety of ways (Box 1), depending in part on the bones affected. Most commonly, the disease involves the pelvis, lumbosacral spine, skull, femur or tibia, but any bone may be affected, and the disease may be localised to one or a few bones or widespread throughout the skeleton. Not infrequently, the disease is asymptomatic, and is detected as an incidental finding at radiography or because of an increase in serum alkaline phosphatase activity.
Pagetic bone pain occurs in a minority of patients. Sometimes this has distinct characteristics, being constant (day and night), present at rest and poorly localised, with a dull, boring character. Other patients have more localised pain, worse on weight-bearing and sometimes self-limiting, which may arise from microfractures or localised lytic lesions. Paget’s disease adjacent to a joint can cause secondary arthropathy with the clinical features of osteoarthritis. Deformity, such as bowing of the femur or tibia, may be asymptomatic or associated with mechanical pain in the affected limb or on the contralateral side, arising from secondary gait problems. Cortical fissure fractures arise from abnormal mechanical forces on weakened bone. They may be asymptomatic or painful. They often remain unchanged over time with no response to treatment, but can progress to complete fracture (Box 2).
The most common neurological complication is deafness arising from involvement of the petrous temporal bone. It may be conductive in nature (from involvement of the middle-ear ossicles), sensorineural (from auditory nerve compression or cochlear involvement) or mixed. Other neurological syndromes are uncommon, but include vertigo, spinal cord compression, local compression syndromes, such as cranial nerve palsies, and, rarely, hydrocephalus or brainstem compression from basilar invagination. Malignant transformation of pagetic bone to osteosarcoma is rare (lifetime risk, < 1%).
Diagnostic imaging
Paget’s disease is diagnosed primarily by radiological examination. Early in the course of the disease, lytic activity predominates, causing focal osteolytic lesions (osteoporosis circumscripta) or flame-shaped, advancing lytic wedges in the long bones (Box 2A). Subsequently, areas of sclerosis develop, leading to the characteristic appearances of mixed lytic and sclerotic areas, thickened trabeculae, bone expansion, cortical thickening and deformity. The radiological appearances are usually characteristic, but occasionally a differential diagnosis of sclerotic or lytic metastases needs to be considered. An isotope bone scan is recommended in all patients as part of the initial diagnostic assessment to determine the distribution of the disease, in particular the involvement of sites with the potential for complications, such as base of skull, spine and long bones (Box 3). Computed tomography scanning is helpful to assess skull-base involvement (including patients with deafness), spinal stenosis or other neurological complication.
Biochemistry
Plasma total alkaline phosphatase level is the most clinically useful marker of disease activity. In patients with monostotic or limited disease, the level may be in the upper reference range: thus, a normal alkaline phosphatase level does not exclude the disorder. Liver function tests should be performed, in particular measurement of γ-glutamyltranspeptidase level, as liver disease can also result in elevated alkaline phosphatase activity, causing diagnostic confusion (Box 1). If there is doubt as to the tissue origin of elevated levels of plasma alkaline phosphatase, assessment of alkaline phosphatase isoenzymes should be requested. In patients whose total alkaline phosphatase level is not elevated, and in those with liver disease, measurement of bone-specific alkaline phosphatase level (where available) or urine markers of bone resorption is helpful. Biochemical assessment should also include measurement of 25-hydroxyvitamin D for two reasons: osteomalacia can present with bone pain and a raised alkaline phosphatase level, and vitamin D deficiency should be corrected before prescribing bisphosphonate therapy.
Treatment
Paget’s disease is treatable, but not all patients require treatment (see Box 4). Bone pain at pagetic sites is an indication for treatment, as there is good evidence that this responds to bisphosphonate therapy.3 In individual patients, it can be difficult to determine whether pain is arising from Paget’s disease or from coexisting conditions such as osteoarthritis. In such cases, a therapeutic trial of bisphosphonate therapy is reasonable if the pain localises to an area of pagetic involvement. Patients with neurological complications should receive treatment, particularly those with spinal stenosis or cord compression, as symptoms may improve with medical therapy.3
Currently, only patients with symptomatic Paget’s disease are eligible for Pharmaceutical Benefits Scheme (PBS) subsidies for their treatment. A strong case can also be made for treating asymptomatic patients in whom the location of the disease puts them at risk of future complications. This includes those with involvement of the long bones such as femur, tibia or humerus, particularly if significant lytic lesions are present, and those with vertebral involvement, because of the risk of fracture or spinal stenosis. Many authorities regard involvement of the base of the skull as an indication for treatment, because of the risk of pagetic deafness, which, once present, rarely responds to medical or surgical therapy. It may also be reasonable to treat younger patients with asymptomatic involvement of joint surfaces, such as the acetabulum, in the hope of preventing secondary arthritis. Evidence that treatment prevents pagetic complications is limited, and unlikely ever to be available from randomised controlled trials. In addition, preoperative medical treatment is recommended for patients undergoing elective surgery (such as joint replacement) to bones with metabolically active Paget’s disease. The rationale for this is that bisphosphonate treatment reduces skeletal blood flow in Paget’s disease,16 and therefore may reduce intraoperative blood loss. Direct evidence for reduced blood loss is, however, lacking.2
Potent second- and third-generation bisphosphonates are the treatment of choice for Paget’s disease, and have superseded other treatments (such as calcitonin and the first-generation bisphosphonate etidronate). In Australia, the most widely used drugs are pamidronate, alendronate and risedronate (Box 4).
Pamidronate is administered by slow intravenous infusion over 2–4 hours. A variety of treatment regimens have been used.2,3 A single dose of 60 mg is adequate for some patients with mild disease,2,17 whereas two to four doses (and occasionally more), administered days or weeks apart, are required for more severe disease.
Alendronate is conventionally prescribed as a 6-month treatment course,2,3,18,19 but recent data suggest that 3 months’ treatment is adequate for many patients.17,20
Risedronate is prescribed as a 2-month course of treatment.
All three drugs are generally well tolerated. The most common side effects of pamidronate are a transient “flu-like” syndrome of fever, myalgia and arthralgia. Serious side effects are rare, but uveitis and acute renal failure have been reported. With alendronate and risedronate, the most common side effects are upper gastrointestinal symptoms. Preliminary data suggest that another intravenous bisphosphonate, zoledronic acid, is also effective for Paget’s disease, and the results of randomised controlled trials are awaited.
Vitamin D deficiency should be corrected with ergocalciferol or cholecalciferol before starting bisphosphonate treatment, and calcium supplementation is advisable to minimise bisphosphonate-induced hypocalcaemia and secondary hyperparathyroidism.21
The aims of treatment are to achieve clinical remission (relief of symptoms), biochemical remission (return of plasma alkaline phosphatase level to the reference range) and radiological remission (filling-in of osteolytic lesions). Disease activity is readily monitored by measuring plasma alkaline phosphatase level every 3 months after starting treatment, and treatment can be stopped when the level returns to the reference range, or when there has been no further reduction on successive measurements. In patients whose alkaline phosphatase level is not elevated at baseline and in patients with liver disease, treatment response can be monitored by observing changes in alkaline phosphatase level within the reference range, measurement of bone-specific alkaline phosphatase or urine markers of bone resorption, or repeat isotope bone scans.
After remission of Paget’s disease has been achieved, patients no longer receiving treatment should be followed up periodically, for example by annual clinical review, with measurement of alkaline phosphatase level. For patients with osteolytic lesions at baseline, repeat radiography should also be performed. Repeat isotope bone scanning is rarely required, as the distribution of affected bones does not change over time, and disease activity can be assessed by simpler means. Relapse inevitably occurs, often several years after the initial treatment course, and patients should then be offered further therapy. Relapse is usually apparent from an increase in alkaline phosphatase activity, but in some patients clinical relapse (recurrent pain) and radiological relapse (progression of osteolytic lesions) precede biochemical relapse. In this case further treatment should be given, even when the alkaline phosphatase level is normal. With repeated courses of treatment the disease can become resistant to individual bisphosphonates. This was first described with etidronate,22 and, more recently, with pamidronate.17,23,24 In such cases, the disease still responds to other potent bisphosphonates; for example, patients whose disease has become resistant to pamidronate can be effectively treated with alendronate or risedronate.17,23
Non-pharmacological treatments should not be neglected. For example, patients with deformity resulting in leg shortening may benefit from shoe raises or orthotics to assist with pain relief and gait difficulties.
Conclusion
Paget’s disease is a common, treatable disorder affecting older Australians. Further research into the genetics of this disease will lead to a better understanding of its pathogenesis, and potentially to new treatments.
1 Clinical presentation and investigation of Paget’s disease
Clinical presentation |
Investigation |
||||||||||||||
|
|
||||||||||||||
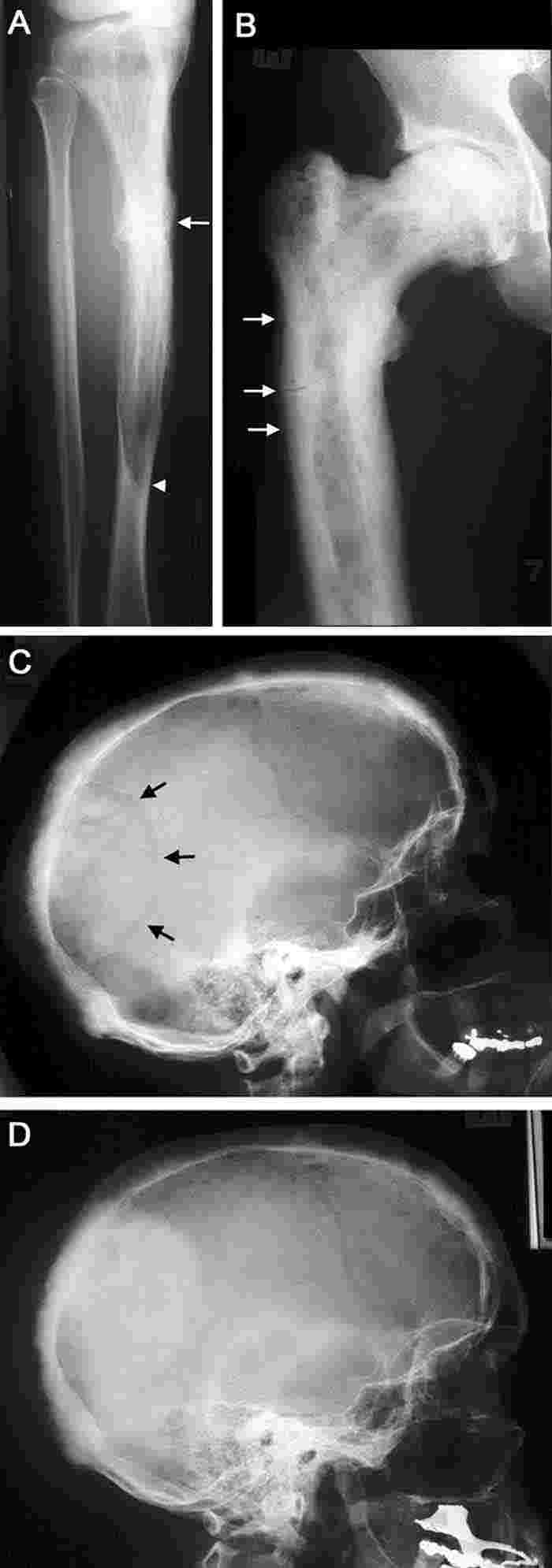
2 Radiographic features of Paget’s disease
A. Paget’s disease of the tibia, with a flame-shaped lytic wedge (arrowhead) and a pathological fracture (arrow).
B. Longstanding Paget’s disease of the femur, with bone expansion, trabecular thickening, mixed lytic and sclerotic areas, and fissure fractures (arrows).
C. Active Paget’s disease of the skull, with marked cortical thickening and an area of osteoporosis circumscripta (arrows).
D. The same patient some years later (after bisphosphonate treatment), with the lytic lesion largely replaced by sclerotic bone.
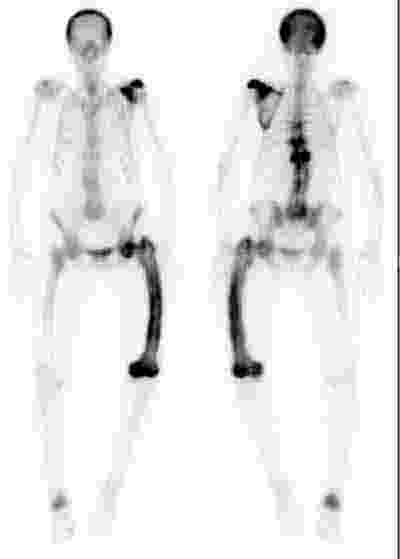
3 Isotope bone scanning in Paget’s disease
A patient with polyostotic Paget’s disease, showing involvement of the skull, left scapula, thoracic vertebrae (T7, T9 and T11), left femur and right calcaneum.
4 Indications for bisphosphonate therapy and the drugs recommended
Indications
Pain in pagetic bones or joints
Neurological complications
Significant osteolytic lesions with fracture risk
Involvement of long bones, vertebrae or base of skull
Patients undergoing surgery to pagetic bones
Asymptomatic joint involvement
Recommended drugs
Pamidronate — 60 mg, given intravenously, 1–4 doses, days or weeks apart
Alendronate — 40 mg/d, given orally for 3–6 months
Risedronate — 30 mg/d, given orally for 2 months
Competing interests
I have received travel assistance from Merck Sharp & Dohme to attend a meeting, and funding from Merck Sharp & Dohme and Novartis for clinical trials in Paget’s disease.
Acknowledgements
I would like to thank the Audiovisual Production Unit and the Nuclear Medicine Department at Sir Charles Gairdner Hospital for assistance with the illustrations.
References
- Paget J. On a form of chronic inflammation of bones. Medico-chirurgical Transactions 1877; 65: 37-63. i1085738
- Lyles KW, Siris ES, Singer FR, Meunier PJ. A clinical approach to diagnosis and management of Paget’s disease of bone. J Bone Miner Res 2001; 16: 1379-1387. i1085740
- Selby PL, Davie MW, Ralston SH, Stone MD. Guidelines on the management of Paget’s disease of bone. Bone 2002; 31: 366-373. i1085742
- Cooper C, Dennison E, Schafheutle K, et al. Epidemiology of Paget’s disease of bone. Bone 1999; 24: 3S-5S. i1085744
- Gardner MJ, Guyer PB, Barker DJP. Radiological prevalence of Paget’s disease of bone in British migrants to Australia. BMJ 1978; 1: 1655-1657. i1085746
- Cooper C, Schafheutle K, Dennison E, et al. The epidemiology of Paget’s disease in Britain: is the prevalence decreasing? J Bone Miner Res 1999; 14: 192-197. i1085748
- Doyle T, Gunn J, Anderson G, et al. Paget’s disease in New Zealand: evidence for declining prevalence. Bone 2002; 31: 616-619. i1085750
- Siris ES, Ottman R, Flaster E, Kelsey JL. Familial aggregation of Paget’s disease of bone. J Bone Miner Res 1991; 6: 495-500. i1085752
- Seton M, Choi HK, Hansen MF, et al. Analysis of environmental factors in familial versus sporadic Paget’s disease of bone — the New England Registry for Paget’s Disease of Bone. J Bone Miner Res 2003; 18: 1519-1524. i1085754
- Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 2002; 70: 1582-1588. i1085756
- Hocking LJ, Lucas GJ, Daroszewska A, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet 2002; 11: 2735-2739.
- Hocking LJ, Lucas GJA, Daroszewska A, et al. UBA domain mutations of SQSTM1 in Paget’s disease of bone: genotype phenotype correlation, functional analysis and structural consequences. J Bone Miner Res 2004; 19: 1122-1127. i1085759
- Duran A, Serrano M, Leitges M, et al. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev Cell 2004; 6: 303-309. i1085761
- Suda T, Takahashi N, Udagawa N, et al. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev 1999; 20: 345-357. i1085763
- Helfrich MH, Hobson RP, Grabowski PS, et al. A negative search for a paramyxoviral etiology of Paget’s disease of bone: molecular, immunological, and ultrastructural studies in UK patients. J Bone Miner Res 2000; 15: 2315-2329. i1085765
- Walton KR, Green JR, Reeve J, Wootton R. Reduction of skeletal blood flow in Paget’s disease with disodium etidronate therapy. Bone 1985; 6: 29-31. i1085767
- Walsh JP, Ward LC, Stewart GO, et al. A randomized clinical trial comparing oral alendronate and intravenous pamidronate for the treatment of Paget’s disease of bone. Bone 2004: 34: 747-754. i1085769
- Siris E, Weinstein RS, Altman R, et al. Comparative study of alendronate versus etidronate for the treatment of Paget’s disease of bone. J Clin Endocrinol Metab 1996; 81: 961-967. i1085771
- Reid IR, Nicholson GC, Weinstein RS, et al. Biochemical and radiologic improvement in Paget’s disease of bone treated with alendronate: a randomized, placebo-controlled trial. Am J Med 1996; 101: 341-348. i1085773
- Khan SA, Vasikaran S, McCloskey EV, et al. Alendronate in the treatment of Paget’s disease of bone. Bone 1997; 20: 263-271. i1085775
- Stewart GO, Gutteridge DH, Price RI, et al. Prevention of appendicular bone loss in Paget’s disease following treatment with intravenous pamidronate disodium. Bone 1999; 24: 139-144. i1085777
- Altman RD. Long-term follow-up of therapy with intermittent disodium etidronate in Paget’s disease of bone. Am J Med 1985; 79: 583-590. i1085779
- Gutteridge DH, Ward LC, Stewart GO, et al. Paget’s disease: acquired resistance to one aminobisphosphonate with retained response to another. J Bone Miner Res 1999; 14 Suppl 2: 79-84. i1085781
- Trombetti A, Arlot M, Thevenon J, et al. Effect of multiple intravenous pamidronate courses in Paget’s disease of bone. Rev Rhum Engl Ed 1999; 66: 467-476. i1085783