





Volume 179 - Issue 8
Adverse event reporting in clinical trials: room for improvement
Authors: Winston S Liauw and Richard O Day
Med J Aust 2003; 179 (8): 426-428. || doi: 10.5694/j.1326-5377.2003.tb05621.x
Published online: 20 October 2003
Published online: 20 October 2003
Abstract
Regulatory and ethical guidelines require clinical trial sponsors to disseminate clinical trial adverse event reports to involved investigators and human research ethics committees.
Compliance with these guidelines has resulted in a major administrative burden for ethics committees.
This burden does not necessarily contribute to the protection of clinical trial participants.
Rationalisation of the adverse event reporting might allow better use of the data and might benefit human research ethics committees.
Good clinical practice requires clinical trial investigators to report serious adverse events (any untoward medical occurrence that results in hospitalisation or prolongation of hospitalisation, or is life-threatening, or results in death or disability, including birth defects) to clinical trial sponsors (see Box), who then have a responsibility to notify all study investigators or trial sites of these events.1 In turn, the institutional human research ethics committee (HREC) is notified by the investigator, and the HREC confirms that it has been notified and advises the sponsor, via the investigator, of any action it mandates. Sponsors are required to report to the Therapeutics Goods Administration (TGA) all individual adverse events (defined as any untoward medical occurrences in patients or clinical investigation subjects, temporally associated with the use of a medicinal product, whether or not considered related to the medicinal product) recorded in Australia, but not events that occur overseas.2 Furthermore, the TGA requires sponsors to report any significant safety concerns or actions taken as a result of analysis of adverse event reports within Australia and overseas to investigators and HRECs, and the TGA undertakes to ensure that this occurs.2 These requirements have evolved over recent years, and have led to a substantial increase in interactions between sponsors, investigators, HRECs, clinical trial monitors, and regulatory agencies, often with multiple interactions related to one adverse event. We are concerned that this increase in activity has not enhanced safety for participants in clinical trials.
From a regulatory perspective, this process effectively fulfils the bureaucratic requirement of data dissemination. However, we contend that the mechanisms for disseminating the data lead to inefficient use of resources, do not deliver data in a timely fashion, fail to provide data in an appropriate context, and contribute minimally to protecting clinical research participants. In effect, a costly paper trail provides data rather than information, creating a situation in which there is a real danger of losing the “needle” —important adverse reactions to drugs — in the “haystack” of all the adverse event reports.
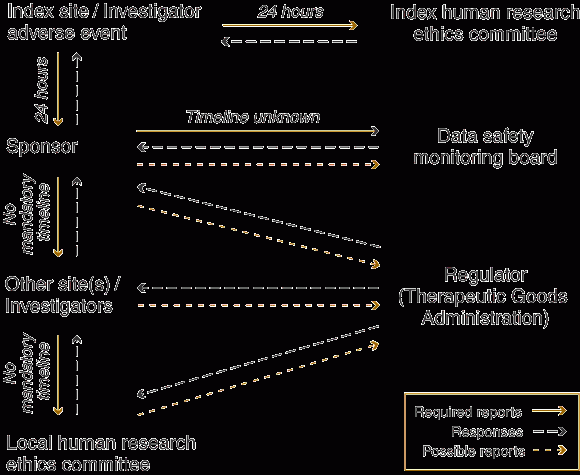
The Box shows the way in which an index site or investigator notifies its local HREC and the sponsor about a serious adverse event. In turn, the sponsor notifies other sites and investigators, who are not necessarily working on the same protocol. Investigators at the other sites assess the report and forward it and their comments to their local HREC. However, the HREC is not in a situation to make adequate use of the report. Firstly, although such reports usually provide substantial detail, significant time has often elapsed since the index event. Also, safety reports do not usually place the adverse event in the wider context of the clinical trial, or of the total experience with the drug. For example, the number of similar adverse events observed, the number of participants accrued, and the total duration of follow-up is not provided to allow the investigator or the HREC to evaluate the significance of the adverse event in an appropriate context.
Because they have access to more complete information, independent data safety monitoring boards are better placed than HRECs to make judgements about safety, particularly in relation to causality.3 It seems obvious that sponsors should take responsibility for establishing effective, independent data safety monitoring boards, as the major liability for adverse events from investigational drug studies lies with the sponsor. It is notable, however, that HRECs have limited ability to assess data safety monitoring boards (eg, how independent they are, if they can act in a timely manner), which act primarily through the sponsor.
Furthermore, while the HREC and investigators currently have a responsibility to review each adverse event report, there is no mechanism to ensure this occurs. Certainly, in the case of non-remunerated, voluntary members of ethics committees, there may simply be insufficient time and resources to undertake such an enterprise when an individual committee is considering dozens of protocols at any one time.3,4
In the administrative situation created by the current process, serious safety concerns may be missed in an ever-growing paper trail. The St Vincent’s Hospital (Darlinghurst) HREC typically receives 80–100 multiple-page reports each month, and review of these reports accounts for roughly half the executive’s review agenda (R Ecclestone, Executive Officer, Research Office, St Vincent’s Hospital, personal communication). Previous review of the role and the workload of Australian HRECs has led to the recommendation for improved resourcing of these bodies.5 Not surprisingly, HRECs have started to look at more systematic and less resource-intensive methods of fulfilling their obligations.
Some consider that HRECs have a duty of care to monitor adverse events reported in a clinical trial so that action can be taken when appropriate. This may well be the case, but it is the sponsor who is liable for the outcome of the adverse event and, ultimately, it is the sponsor who has a duty to provide the most efficient adverse event reporting system to protect trial participants.
Potential solutions
The ready availability of electronic communication services should allow the rapid accumulation of (serious) adverse event data that can be collated and disseminated to the concerned parties. Instead of an exhaustive description of each event, the sponsor could provide investigators with a tabulated summary of all of the adverse events observed in the study, a summary of the numbers and types of events along with the numbers of patients exposed to the drug and the duration of their exposure, and any recommended actions. The frequency of reports might be dictated by the nature of the trial. Small Phase II studies with potentially toxic drugs may need daily summary reports, whereas large Phase III trials incorporating drugs with relatively well characterised toxicity profiles may only need weekly or fortnightly reports. Individual reports could be available on request, possibly through a password-protected website. A centralised system to track local HREC decisions concerning adverse events, as well as other protocol-related matters, has also been proposed.6 Such a system would allow the local HREC access to the decisions generated by the entire body of HRECs overseeing any particular trial.
The utility of an expedited, simplified reporting process is illustrated, in part, by the American North Central Cancer Treatment Group’s Real-Time Toxicity Monitoring Program.7 This program expedited reporting of serious adverse events to a data monitoring group. Over a 3-year period of operation, this led to protocol modifications in six Phase II trials. Given the toxicity associated with cytotoxic drugs, it could reasonably be argued that such a program may not be as relevant to the monitoring of trials in general. However, identifying serious events is not the only important reason for monitoring toxicity. The sponsor and investigators also have a responsibility to inform study participants of the potential risks of participating in the study. A more efficient mechanism for event reporting may improve the informed- consent process.
Centralising components of the functions of HRECs may allow a tier of comprehensive safety review achievable by a data safety monitoring board, but not by individual HRECs.3 Currently, the NSW Department of Health is supporting a pilot program for a Shared Scientific Assessment Scheme (Ainsley Martlew, Secretary, NSW SSAS, personal communication). At this stage, the scheme has not accepted a mandate to oversee safety for trials. However, such a responsibility is conceivable. Certainly, in regulatory environments where independent or contract HRECs exist, such a process is under consideration or being implemented.8
Finally, HRECs should routinely insist that the data safety monitoring board process should be made more transparent. Protocols should clearly state whether or not an independent monitoring board is being established, and the membership of that board and its terms of reference should be disclosed. The sponsor’s internal operating procedure for handling adverse events should also be disclosed. Rationalisation of the HREC adverse event monitoring activities could allow HRECs to ensure that appropriate data safety monitoring board activities are conducted without usurping the power or independence of those committees. HRECs need to be better resourced to allow closer collaboration with investigators to ensure adequate implementation of clinical trials, and monitoring that acts as quality improvement rather than an auditing process.
There is no immediate solution to these issues, but discussion between the stakeholders, including sponsors’ representatives, representatives of the Australian Regulatory and Clinical Scientists, the Australian Health Ethics Committee, consumers (trial participants) and the TGA, is suggested. Good clinical practice requires close scrutiny to ensure that it is not only ethical, but also practical.
Tracking an adverse event
Several deficiencies can be identified in the way adverse events are currently reported:
Mandated timelines exist only for the initial reporting. It may take months for the original event to be reported to additional human research ethics committees.
Each site/investigator is isolated on its own branch of the diagram, with only the sponsor having an overall perspective on the deliberations of all parties.
The activities of the data safety monitoring board are not transparent and the board has little or no direct communication with investigators, ethics committees or regulators.
The number of iterations that a report undergoes (and consequent correspondence to and fro) is not illustrated in the diagram.
There are several tiers of causality assessment — at the level of the index investigator, the data safety monitoring board, the additional sites and, finally, the human research ethics committee. Only the sponsor and data safety monitoring board have access to the complete data.
Competing interests
None identified.
Acknowledgements
We thank Mr Rodney Ecclestone, Executive Officer, St Vincent’s Hospital (Sydney) Human Research Ethics Committee, and Ms Barbara Yeung, Clinical Trials Centre, St Vincent’s Hospital, for helpful discussions concerning the adverse event reporting process.
References
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH harmonised tripartite guideline: guideline for good clinical practice E6, 1996. Available at: www.ich.org/pdfICH/e6.pdf (accessed Aug 2003).
- Human research ethics committees and the therapeutic goods legislation. January 2001. Commonwealth Department of Health and Aged Care, May 2001. Available at: www.health.gov.au/tga/docs/pdf/unapproved/hrec.pdf (accessed Aug 2003).
- Christian MC, Killen J, Abrams JS, et al. A central institutional review board for multi-institutional trials. N Engl J Med 2002; 346: 1405-1408. i1082868
- Burman WJ, Reeves RR, Cohn DL, Schooley RT. Breaking the camel’s back: multicenter clinical trials and local institutional review boards. Ann Intern Med 2001; 134: 152-157. i1082870
- Report of the review of the role and functioning of institutional ethics committees. Report to the Minister for Health and Family Services. Canberra: AGPS, 1996. i1082872
- Ferris LE. Industry-sponsored pharmaceutical trials and research ethics boards: are they cloaked in too much secrecy? CMAJ 2002; 166: 1279-1280. i1082874
- Goldberg RM, Sargent DJ, Morton RF, et al. Early detection of toxicity and adjustment of ongoing clinical trials: the history and performance of the North Central Cancer Treatment Group’s Real-Time Toxicity Monitoring Program. J Clin Oncol 2002; 20: 4591-4596. i1082876
- Institutional Review Boards: the emergence of independent boards. Washington, DC: US Department of Health and Human Services, Office of Inspector General, 1998: 1-18. (Publication No. OEI-01-97-00192). Available at: oig.hhs.gov/oei/reports/oei-01-97-00192.pdf (accessed Aug 2003).