





Volume 177 - Issue 4
Severe congenital lead poisoning in a preterm infant due to a herbal remedy
Authors: Paul A Tait, Amish Vora, Simon James, D James Fitzgerald and Beverly A Pester
Med J Aust 2002; 177 (4): 193-195. || doi: 10.5694/j.1326-5377.2002.tb04731.x
Published online: 19 August 2002
Published online: 19 August 2002
Chronic lead poisoning may manifest clinically as abdominal pain, constipation, proteinuria, haematuria, peripheral neuropathy and muscle weakness.1 With more significant lead poisoning, encephalopathy may occur.2 Sustained blood lead levels of over 0.5 μmol/L in early childhood are likely to be associated with intellectual underperformance.3 However, congenital lead poisoning and its subsequent management has rarely been reported in preterm infants.
Treatment is with chelating agents, which reduce lead concentrations in the blood and tissues by forming water-soluble complexes that are cleared by the kidneys.4
We report a case in which the neonatal blood lead level was the highest recorded for a surviving infant.
Clinical record
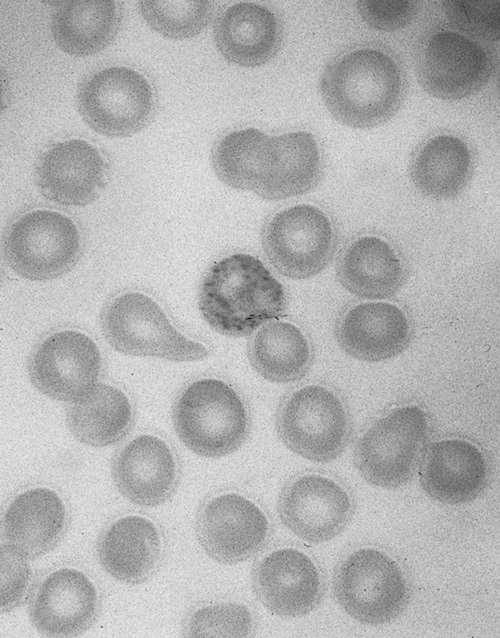
A 24-year-old pregnant woman, who had recently emigrated from India, was found to have a haemoglobin level of 70 g/L at 24 weeks' gestation. At that time it was noted that she was a vegan and had a normal blood film and iron studies. At 30 weeks' gestation, she presented with abdominal pain and a progressive confusional state culminating in seizures. The blood film now showed basophilic stippling, a typical sign of lead poisoning (Box 1), and subsequent testing showed she had a blood lead concentration of 5.2 μmol/L (the National Health and Medical Research Council's public health goal is a level of ≤ 0.48 μmol/L).3
Chelating therapy was initiated with intramuscular dimercaprol and intravenous calcium disodium edetate (CaNa2EDTA).5 Thirty-six hours later the woman had an antepartum haemorrhage, and, after induction of labour, she gave birth to a 1.6 kg (75th percentile) female baby by vaginal delivery. Apgar scores were 4 and 6. The infant was flaccid and areflexic and did not move in response to noxious stimuli, although spontaneous ocular movements were present. As she had emerging alveolar hypoventilation and no gag reflex, she was intubated and ventilated. The clinical suspicion of bilateral diaphragmatic palsy was later confirmed by fluoroscopy.
Basophilic stippling was not seen on the infant's blood films, although Heinz bodies were present. Lead concentration in the cord blood was 7.6 μmol/L (12 hours before birth, maternal blood lead concentration had been 2.3 μmol/L). The concentration of erythrocyte porphyrins was 20.3 μmol/L (normal range, 0.4–1.7 μmol/L), and radiographs of the long bones showed an increase in the bone density adjacent to the metaphyses (both signs of lead poisoning).
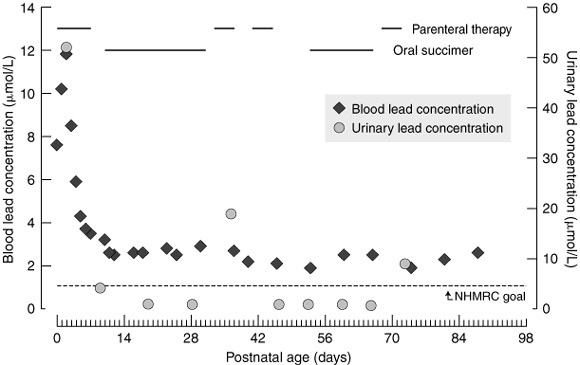
Within 24 hours of birth, the infant was commenced on chelation therapy (intramuscular dimercaprol 4 mg/kg/dose every four hours and intravenous CaNa2EDTA 50 mg/kg/day, given after the second dose of dimercaprol). The blood lead concentration initially rose to 11.8 μmol/L within the first 48 hours of therapy, then fell rapidly over the next few days. The high urinary lead concentration of 52 μmol/L on Day 3 was an indicator that lead was being excreted. The time course of blood and urinary lead concentrations for the first 14 weeks post partum is shown in Box 2. On Day 7, it was judged appropriate to cease intravenous chelating therapy. Succimer (an oral chelating agent) 30 mg/kg/day5 was commenced three days later. At that time, the urinary lead concentration had fallen to 4.2 μmol/L. Over the three-week course of succimer, urinary lead concentrations fell further, while blood lead concentrations remained relatively constant. As the succimer appeared to be having little effect, it was discontinued on Day 31, and intravenous CaNa2EDTA was recommenced 48 hours later. A rise in urinary lead excretion was observed after this course and subsequent courses of parenteral therapy.
By Day 42, facial, bulbar, proximal-limb and diaphragmatic muscle activity had improved sufficiently to allow successful extubation. By Day 53, the blood lead concentration appeared to have fallen to a satisfactory level, and an attempt at maintenance therapy using oral succimer at the higher dose of 60 mg/kg/day was commenced.6,7 However, the blood lead concentration increased during the second course of succimer, with a corresponding decrease in the urinary lead concentration. Intravenous CaNa2EDTA was recommenced. When chelation therapy was subsequently discontinued for two weeks, the blood lead concentration again rose, and a fifth course of CaNa2EDTA was initiated.
Progress of infant after first 14 weeks. By three months' corrected age (ie, about 5 months after birth) the infant was able to fully feed by sucking, although there was significant gastroesophageal reflux. Her peripheral weakness had almost resolved, but bilateral wrist drop and poor head control persisted. Blood lead concentration had fallen to 1.8 μmol/L. It was decided to reintroduce oral succimer at 60 mg/kg/day. This appeared to be effective, as the lead levels not only fell in the blood but increased in the urine.
A brainstem auditory response at four months' corrected age showed right sensorineural deafness. (A magnetic resonance image of the brain at three weeks of age had been normal.) Throughout the hospital stay her serum calcium, zinc, iron, renal and liver function tests were normal.
The infant was discharged home, on succimer, at five months' corrected age with a blood lead concentration of 0.95 μmol/L. A neurodevelopmental examination at the time revealed a two-month delay. Her two siblings, aged two and four years, were found to have blood lead concentrations of 0.6 and 0.3 μmol/L, respectively.
Lead source identification. An environmental audit of the home and the local Sikh community kitchen revealed no obvious source of lead. Interviews conducted with the mother established that she had been taking several tablets, prescribed by an Ayurvedic doctor in India for treatment of a gastrointestinal complaint, periodically over the course of the past nine years. Analytical results for the various tablets are presented in Box 3, the most notable being the high lead content of two of the tablet types (4.5%–8.9% lead). The mother's tablet use in the nine months before the child's birth represented a lead intake of at least 50 times the average weekly lead intake of Western populations.2,8
Correspondence with the Ayurvedic doctor in India, who prescribed the brown ("HSY-15") tablets in this case, has not yet provided any clues as to the origin of the lead in these tablets.
Discussion
Clinical considerations
The diagnosis of intrauterine lead intoxication was made on the basis of a maternal encephalopathy with a high blood lead concentration, maternal anaemia with basophilic stippling, and a high cord-blood lead concentration. Infant blood lead levels of a third to a half the peak level reported here are usually associated with severe cerebral oedema and death.2 In this instance, the infant was encephalopathic and profoundly weak and had diaphragmatic palsy at birth. The limb weakness clinically appeared to be predominantly neuropathic. Several electrophysiological attempts to clarify the presence of either a neuropathy or a myopathy failed owing to technical and interpretive difficulties. Although lead-induced peripheral neuropathy is well described,9 diaphragmatic palsy is unreported. There was no evidence of renal involvement or proteinuria, possibly due to the infant's renal immaturity. There was no basophilic stippling in any neonatal blood film. This relative paradox — the absence of basophilic stippling despite lead intoxication — has been noted previously.10 The unilateral sensorineural deafness is likely to be due to lead toxicity.11
Previous reports of congenital lead intoxication indicate that maternal and cord blood lead levels correlate.12 The marked discrepancy in this case (2.3 μmol/L [mother] v 7.6 μmol/L [cord]) could be due to the possibility that at high maternal blood lead concentrations the lead-binding capacity of the erythrocytes becomes saturated13 and more lead is available for transfer to the fetus. Alternatively, the discrepancy could be due to in-utero mobilisation of lead from fetal tissues without adequate maternal clearance. This latter consideration indicates a potential detrimental effect on the fetus of chelation therapy in pregnancy.
As chelating agents bind lead into complexes that are cleared by the kidneys, an increase in urinary lead concentration was expected after administration of each of the chelating agents. This phenomenon was consistently observed after courses of dimercaprol and CaNa2EDTA. However, variable doses of oral succimer during the preterm period did not lead to corresponding increases in urinary lead concentration, despite raising the dose to a level higher than those recommended in the literature.5-7 Only when oral succimer was recommenced at three months' corrected age did the predicted response occur. This apparent early ineffectiveness of succimer could be due to absorptive failure from the infant's immature gut. Furthermore, succimer is a prodrug that requires cysteine for activation.2 It is unclear whether or not this enzymatic activation process is mature in the premature infant.
Public health considerations
While lead was the principal concern in this case, mercury was also detected in some of the tablets (Box 3). Weekly ingestion of one of the tablets containing 400 μg would exceed the Australian average weekly mercury intake by at least fourfold.8 Mercury poisoning was of secondary concern in the current circumstance, and chelation therapy was expected to remove the mercury along with the lead. This does, however, illustrate that herbal medicines may contain more than one potentially toxic component.
In many countries, including Australia, it is legal to import herbal remedies for personal use, although some restrictions apply.14 In addition, testing programs for contaminants in herbal remedies are virtually non-existent. Acute lead poisoning has been reported from traditional and herbal medicines and cosmetics obtained from India,15-17 the Middle East,18-19 Mexico20 and Asia.20-21 This should be a matter of grave concern to patients who take such remedies, and to health professionals and health authorities in these countries and in Australia.
2: Infant blood and urinary lead concentrations from birth to 14 weeks of age*
*Horizontal lines across top of graph indicate periods of parenteral therapy with calcium disodium edetate and oral succimer. Dotted horizontal line shows National Health and Medical Research Council public health goal for maximum lead concentration in blood (0.48 μmol/L).
Competing interests
None identified.
Acknowledgements
The authors gratefully acknowledge Dr Stephen Rothenberg, of Charles Drew University, Los Angeles, for discussion of fetal and maternal blood lead levels; Mike Stevenson, of Berri/Barmera Council, for the environmental audit; Sam Mangas, for assistance with the source investigation; and Bill Dollman and Dr Brian Priestly, for discussions on the regulation of herbal remedies in Australia.
References
- Fuhrman BP. The acutely ill child. In: Nelson's essentials of paediatrics. 2nd ed. Philadelphia: WB Saunders, 1994: 91-122. i1067487
- Klaassen CD. Heavy metals and heavy-metal antagonists. In: Goodman and Gilman's The pharmacological basis of therapeutics. 10th ed. New York: McGraw Hill, 2001: 1851-1875. i1067489
- Statement issued by 115th session of the National Health and Medical Research Council, 2 June 1993. Revision of the Australian guidelines for lead in blood and lead in ambient air. Available at: <http://www.nhmrc.gov.au/publications/synopses/eh8syn.htm>. Link updated August 2005.
- Parfitt K, editor. Martindale: the complete drug reference. 32nd ed. London: Pharmaceutical Press, 1999. i1067493
- Treatment guidelines for lead exposure in children (RE9529). American Academy of Pediatrics Committee on Drugs. Pediatrics 1995; 96: 155-160. i1067495
- Graziano JH, Lolacono NJ, Meyer P. Dose-response study of oral 2,3-dimercaptosuccinic acid in children with elevated blood lead concentrations. J Pediatr 1988; 113: 751-757. i1067497
- Graziano JH, Lolacono NJ, Moulton T, et al. Controlled study of 2,3-dimercaptosuccinic acid for management of childhood lead intoxication. J Pediatr 1992; 120: 133-139. i1067499
- 19th Australian Total Diet Survey. A total diet survey of pesticide residues and contaminants. Canberra: Australian New Zealand Food Authority, 2001. i1067501
- Howard H. Heavy metal poisoning. In: Harrison's principles of internal medicine. 14th ed. New York: McGraw-Hill; 1998: 2564-2569. i1067503
- Timpo AE, Amin JS, Casalino MB, Yuceoglu AM. Congenital lead intoxication. J Pediatr 1979; 94: 765-767. i1067505
- Knecht CD, Crabtree J, Katherman A. Clinical, clinicopathologic, and electroencephalographic features of lead poisoning in dogs. J Am Vet Med Assoc 1979; 175: 196-201. i1067507
- Goyer RA. Transplacental transport of lead. Environ Health Perspect 1990; 89: 101-105. i1067509
- Marcus AH. Multicompartment kinetic model for lead. III. Lead in blood plasma and erythrocytes. Environ Res 1985; 36: 473-489. i1067511
- Therapeutic Goods Administration. Australian Department of Health and Ageing. 2002. Available at: <http://www.tga.gov.au/cm/cm.htm>. Link updated August 2005.
- McElvaine MD, Harder EM, Johnson L, et al. Lead poisoning from the use of Indian folk medicines. JAMA 1990; 264: 2212-2213. i1067515
- Dunbabin DW, Tallis GA, Popplewell PY, Lee RA. Lead poisoning from Indian herbal medicine (Ayurveda). Med J Aust 1992; 157: 835-836.
- van Vonderen MGA, Klinkenberg-Knol EC, Craanen ME, et al. Severe gastrointestinal symptoms due to lead poisoning from Indian traditional medicine. Am J Gastroenterol 2000; 95: 1591-1592. i1067518
- Shaltout A, Yaish SA, Fernando N. Lead encephalopathy in infants in Kuwait. Ann Trop Paediatr 1981; 1: 209-215. i1067520
- Rahman H, Al Khayat A, Menon N. Lead poisoning in infancy – unusual causes in the U.A.E. Ann Trop Paediatr 1986; 6: 213-217. i1067522
- Lead poisoning associated with use of traditional ethnic remedies — California, 1991–1992. MMWR Morb Mortal Wkly Rep 1993; 42: 521-524. i1067524
- Adult lead poisoning from an Asian remedy for menstrual cramps — Connecticut, 1997. MMWR Morb Mortal Wkly Rep 1999; 48: 27-29. i1067526