




Marfan syndrome is a genetic condition affecting connective tissue, with a prevalence of at least one per 5000. The major cause of mortality is dissection of a dilated ascending aorta, but other cardiovascular, ocular and musculoskeletal manifestations are common.1 These include subluxation of the ocular lens, overgrowth of the long bones, and other skeletal abnormalities. Marfan syndrome is caused by mutation of the FBN1 gene on chromosome 15, which encodes fibrillin 1, a large extracellular matrix protein that provides stretch and elasticity to connective tissues and is also involved in regulating the bioavailability of transforming growth factor beta (TGFB) family members.
Clinical criteria for diagnosing Marfan syndrome were developed by an international group in 1996.2 A guide to these 1996 criteria highlighted some of the diagnostic difficulties, including differential diagnoses such as homocystinuria, phenotypic variability within families, other conditions that share some features of Marfan syndrome and issues raised by DNA testing.1 Revised diagnostic guidelines were released in 2010.3 The revised criteria classified 83% of patients with a mutation in FBN1 as having Marfan syndrome, compared with 89% according to the older criteria.4 There are a number of conditions of the connective tissue with a similar phenotype that can be confused with Marfan syndrome, and the recently published modifications of the diagnostic criteria facilitate the differentiation of Marfan syndrome from these conditions.1 We now outline the 2010 revised criteria and how they differ from the previous criteria, as well as other developments in the understanding of Marfan syndrome.
The new criteria are outlined in Box 1, which provides a decision tree to assist in determining whether a patient has Marfan syndrome. In this modified diagnostic process, the focus has shifted from the musculoskeletal signs to the cardiovascular and ocular abnormalities. Thus, the presence of a dilated aorta plus ectopia lentis is now sufficient to give an unequivocal diagnosis of Marfan syndrome. In the previous criteria, involvement of a third system or the patient having an affected family member was also required for diagnosis. In patients with either a dilated aorta or ectopia lentis, the presence of a causal FBN1 mutation is now sufficient to make a diagnosis of Marfan syndrome.
Dilatation of the aorta is determined at the sinus of Valsalva and ascending aorta using charts of the range of normal aorta size in relation to body surface area. Measurements must be corrected for age and body size and compared with normative data using a z-score calculation. Reference graphs and z-score calculations are available.5
In the revised criteria, skeletal, skin, lung, facial, other ocular and other cardiovascular signs have been included in a new scoring system, which resolves some difficulties encountered with the earlier criteria (Box 2). For example, the 1996 criteria included “pectus excavatum requiring surgery” as part of the major skeletal criterion. However, the requirement for surgery is frequently a personal or cosmetic decision, rather than a clinical one. In the revised criteria, any form of pectus excavatum or chest wall asymmetry is allocated one point, regardless of severity, while pectus carinatum is allocated two points since it is less common in the general population and is more frequently associated with Marfan syndrome. The assessment of upper to lower segment ratio has been simplified by providing different threshold values based on age and ethnicity. Pes planus with hindfoot deformity (medial displacement of the medial malleolus) is allocated more points than pes planus without hindfoot deformity, since the latter is frequent in the general population. Two points are allocated to dural ectasia as well as to protrusio acetabulae. For the ocular system, there is no longer a requirement to measure axial length of the globe and shape of the cornea. Myopia of more than 3 dioptres is allocated one point. Mitral valve prolapse is also allocated one point, and other cardiovascular manifestations (eg, aortic aneurysm at other sites) are no longer included.
In this new scoring system, a score of seven points is associated with a diagnosis of Marfan syndrome if there is either aortic dilatation and/or dissection, or a family history of Marfan syndrome (Box 1). Patients who are allocated three points on initial examination should have a pelvic x-ray to check for protrusio acetabulae (two points), and if results are positive, magnetic resonance imaging (MRI) of the spine should be conducted to check for dural ectasia (two points). Patients who are allocated five points on initial examination should have MRI of the spine and/or a pelvic x-ray.
There are now a number of mouse models of Marfan syndrome6 that have been used to understand the natural history of the condition and to evaluate possible treatments. One mouse model showed the critical role of abnormal TGFB signalling in the cardiovascular and lung pathology of Marfan syndrome.7 Fibrillin 1 appears to be involved in regulating bioavailability of TGFB family members. Mutation in FBN1 allows a higher level of active TGFB, a protein involved in immune function, cell proliferation and the epithelial-to-mesenchyme transition. TGFB receptor genes (TGFBR1 and TGFBR2) are mutated in Loeys–Dietz syndrome, a condition that shows phenotypic overlap with Marfan syndrome.8 Recent studies of the angiotensin receptor antagonist losartan, which blocks TGFB signalling, demonstrated significant reduction in cardiovascular and lung abnormalities in a mouse FBN1 mutant strain.9 However, losartan had no effect on the skeletal phenotype,10 whereas the osteoporosis drug alendronate was effective in reducing these signs. Several small clinical trials of beta blockers, angiotensin-converting enzyme inhibitors and angiotensin-II receptor antagonist drugs have shown varying beneficial effects. Currently, losartan, beta blockers and angiotensin-converting enzyme inhibitors are being evaluated in large randomised clinical trials.
A number of other conditions share some features with Marfan syndrome.11 These include related cardiovascular conditions, such as various forms of familial aortic aneurysm disease, bicuspid aortic valve and Loeys–Dietz syndrome. In children, the cardiovascular and skeletal features of Marfan syndrome may not be obvious, and diagnoses of homocystinuria or Klinefelter syndrome may be considered. Applying the revised diagnostic criteria should allow most differential diagnoses to be distinguished. For example, a common diagnosis in our Marfan clinic (28 of 198 index cases) is familial aortic dilatation and/or dissection. The new criteria outlined in Box 1 clearly show that this is not Marfan syndrome unless there is concomitant ectopia lentis, a systemic score of ≥ 7, a known FBN1 mutation or a family history of Marfan syndrome.
In addition, the range of phenotypes caused by mutations in the FBN1 gene has expanded over the last few years.11 For example, FBN1 mutation associated with dilated cardiomyopathy and concomitant risk of sudden death has been increasingly recognised.12 Some FBN1 mutations are associated with phenotypes that contrast with Marfan syndrome, including short stature. This shows that detection of an apparently pathogenic mutation in FBN1 does not necessarily lead to a diagnosis of Marfan syndrome.
DNA testing for FBN1 mutations has been difficult because of the size of the gene (among the largest in the human genome at over 200 000 bases and 65 exons) and because there are few recurrent mutations of FBN1, so novel mutation must be identified for most families. With decreasing costs, it has become feasible to sequence DNA of the whole coding region to detect potentially pathogenic variants. New technical developments in high-throughput sequencing strategies may ultimately assist with mutation discovery.13,14 It is now possible to sequence the exons of all human genes in a single reaction (exome sequencing15), and this approach is being offered commercially for screening for mutations in BRCA1 and BRCA2 genes, which convey susceptibility to hereditary breast and ovarian cancer.16 Since most mutations that result in Marfan syndrome are missense or nonsense mutations of the coding sequence,17 exon sequencing has recently been evaluated as an efficient method for finding disease-associated mutations.18 Capture of the complete FBN1 sequence, including the 5' promoter region, introns and 3' untranslated region, allowed high-throughput sequencing of the whole of the FBN1 gene so that intronic and regulatory mutations were not missed. In addition, the development and decreasing cost of RNA sequencing19 could facilitate sequencing of FBN1 transcripts using RNA from appropriate cell types (for example fibroblasts derived from skin biopsy or aortic smooth muscle cells). RNA sequencing would also allow discovery of abnormal transcription start sites, splice variants and microRNA differences that may affect FBN1 mRNA levels or function. These strategies would capture most mutations but are unlikely to reduce the cost of finding a mutation at present.
Laboratories offering DNA testing for FBN1 mutations in Australia can be found in the Royal College of Pathologists of Australasia Catalogue of Genetic Tests and Laboratories20 and a list of worldwide laboratories is also available.21 When a diagnosis of Marfan syndrome is suspected, the initial clinical evaluation should include cardiovascular, skeletal and ocular examination and assessment of family history. The need for DNA testing and the nature of that testing can be best determined after consultation with a clinical geneticist who will liaise with the laboratory and provide appropriate counselling.
A DNA variant identified in an affected person may or may not be the cause of the condition. In assessing this possibility, the following considerations are important:
Has the DNA change been seen before in a patient with Marfan syndrome or in members of the patient’s family?
What is the frequency of the DNA change in the general population? Variants that are also found in phenotypically normal individuals are unlikely to be causative.
Does the DNA change segregate with the disease in the family? A causative mutation should be found in all affected individuals and be absent in their unaffected relatives.
Would the DNA change alter an amino acid in the protein? If so, is the affected amino acid highly conserved?
Would the DNA change result in premature termination of the protein resulting from replacement of an amino acid codon with a stop codon (so that protein synthesis terminates at that point) or a frame shift (when bases are deleted or inserted in numbers that are not multiples of three)?
Is a large segment of the DNA (or the whole gene) deleted?
Once a putative causative mutation has been identified, family members can be screened to see who has the same mutation. Where no causative variant is found, it may be possible to use a haplotype that includes genetic variants within and close to the FBN1 allele (and also the disease mutation) as a marker for the disease mutation, provided there are at least six informative individuals in the family to confirm linkage.3
Where one member of a couple has Marfan syndrome, involvement of genetic counselling services can ensure that appropriate genetic testing is available to plan pregnancies. Where the family has not yet been evaluated, a mutation must first be identified in the index case, and this takes about 2 months.20 Once the mutation is known, testing of embryos or fetuses can be done quickly. This allows testing of the offspring through amniocentesis, chorionic villus sampling or after in-vitro fertilisation (when a cell of the embryo can be tested before implantation). However, the phenotype in Marfan syndrome is very variable. Some genotype–phenotype correlations have been documented. For example, mutations in central exons tend to be associated with severe Marfan syndrome while mutations at the C-terminal end of the protein may be associated with ectopia lentis, or with Weill–Marchesani syndrome. Therefore, the nature of the mutation provides some predictive information about the severity, prognosis and quality of life of the person who carries it, but there is considerable variation even for those within a family who carry the same FBN1 mutation.22
The revised clinical criteria have simplified the diagnosis of Marfan syndrome by removing subjective criteria and simplifying the assessment of the contribution of signs and symptoms in diverse organ systems. Additional advances in technology and understanding of Marfan syndrome and related conditions will affect patients in a number of ways. As mutation screening and testing become more prevalent, at-risk individuals in families will be identified and appropriate clinical monitoring can be put in place. Although we cannot yet predict the severity of the condition in individual family members, we are beginning to understand the genes that interact with FBN1 and may modify the effect of FBN1 mutation. In the future, DNA analysis of the family of an individual with Marfan syndrome may involve genotyping genes in addition to FBN1. It remains important to assess the families of people with Marfan syndrome in a multidisciplinary clinic so that the differential diagnoses can be fully canvassed and the best possible care and follow-up assured.
1 Decision tree for diagnosing Marfan syndrome using revised criteria3
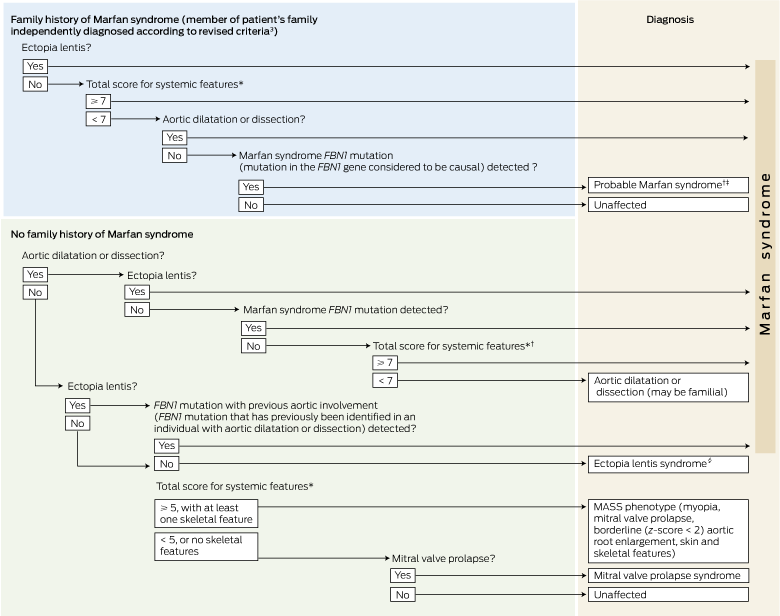
FNB1 = gene encoding fibrillin 1.
* For scoring of systemic features, see Box 2.† In the absence of ectopia lentis or a Marfan syndrome FBN1 mutation, the diagnosis of Marfan syndrome may only be made in the absence of discriminating features of Shprintzen–Goldberg syndrome, Loeys–Dietz syndrome and vascular Ehlers–Danlos syndrome, and after testing for genes encoding transforming growth factor beta receptors (TGFBR2, TGFBR1) and collagen type III alpha 1 (COL3A1) mutations, and for abnormalities of collagen biochemistry. ‡ In individuals with a family history of Marfan syndrome, the presence of the familial FBN1 mutation indicates a high risk of developing clinical evidence of Marfan syndrome. These individuals need regular monitoring, particularly for cardiovascular and ocular signs. § Ectopia lentis syndrome is diagnosed regardless of systemic score and with a novel FBN1 mutation not known to be associated with Marfan syndrome or aortic dilatation, or without FBN1 mutation.
2 Scoring of systemic features for the diagnosis of Marfan syndrome*
Feature |
Score |
||||||||||||||
Wrist OR thumb sign† |
1 |
||||||||||||||
Wrist AND thumb signs† |
3 |
||||||||||||||
Pectus carinatum deformity |
2 |
||||||||||||||
Hindfoot deformity |
2 |
||||||||||||||
Plain pes planus |
1 |
||||||||||||||
Pectus excavatum or chest asymmetry |
1 |
||||||||||||||
Pneumothorax |
2 |
||||||||||||||
Dural ectasia |
2 |
||||||||||||||
Protrusio acetabulae |
2 |
||||||||||||||
Reduced upper segment to lower segment ratio,‡ AND increased ratio of arm span to height§ AND no severe scoliosis |
1 |
||||||||||||||
Scoliosis or thoracolumbar kyphosis |
1 |
||||||||||||||
Reduced elbow extension |
1 |
||||||||||||||
Three of the five typical facial features (dolichocephaly, enophthalmos, downward slanting palpebral fissures, malar hypoplasia, retrognathia) |
1 |
||||||||||||||
Skin striae |
1 |
||||||||||||||
Myopia of > 3 dioptres |
1 |
||||||||||||||
Mitral valve prolapse |
1 |
||||||||||||||
* Many of these features are also illustrated in a previous clinical update.1 † The thumb sign is positive when the entire distal phalanx of the adducted thumb extends beyond the ulnar border of the palm with or without the assistance of the patient or examiner to achieve maximal adduction. The wrist sign is positive when the tip of the thumb overlaps the entire fingernail of the fifth finger when wrapped around the contralateral wrist.1,3 ‡ For white adults, an upper segment to lower segment ratio of < 0.85; for black adults, < 0.78; for children, < 1 at 0–5 years, < 0.95 at 6–7 years, < 0.9 at 8–9 years and < 0.85 at > 10 years. § For adults, a ratio of arm span to height > 1.05. From Loeys et al.3 Reproduced with permission. |
|||||||||||||||
Provenance: <p>Not commissioned; externally peer reviewed.</p>
- Kim M Summers1
- Jennifer A West2
- Annette Hattam3
- Denis Stark4
- James J McGill5
- Malcolm J West2
- 1 The Roslin Institute and Royal (Dick) School of Veterinary Studies, University of Edinburgh, Edinburgh, UK.
- 2 School of Medicine, University of Queensland, Brisbane, QLD.
- 3 Genetic Health Queensland, Queensland Health, Brisbane, QLD.
- 4 Prince Charles Hospital, Brisbane, QLD.
- 5 Department of Metabolic Medicine, Royal Children’s Hospital, Brisbane, QLD.
We are grateful for the support of the Heart Foundation and the Prince Charles Hospital Foundation. The Roslin Institute is supported by an Institute Strategic Programme Grant from the Biotechnology and Biological Sciences Research Council, United Kingdom.
No relevant disclosures.
- 1. Summers KM, West JA, Peterson MM, et al. Challenges in the diagnosis of Marfan syndrome. Med J Aust 2006; 184: 627-631. <MJA full text>
- 2. De Paepe A, Devereux RB, Dietz HC, et al. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 1996; 62: 417-426.
- 3. Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010; 47: 476-485.
- 4. Faivre L, Collod-Beroud G, Adès L, et al. The new Ghent criteria for Marfan syndrome: what do they change? Clin Genet 2012; 81: 433-442.
- 5. Roman MJ, Devereux RB, Kramer-Fox R, O’Loughlin J. Two-dimensional echocardiographic aortic root dimensions in normal children and adults. Am J Cardiol 1989; 64: 507-512.
- 6. Charbonneau NL, Carlson EJ, Tufa S, et al. In vivo studies of mutant fibrillin-1 microfibrils. J Biol Chem 2010; 285: 24943-24955.
- 7. Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet 2003; 33: 407-411.
- 8. Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 2005; 37: 275-281.
- 9. Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006; 312: 117-121.
- 10. Nistala H, Lee-Arteaga S, Carta L, et al. Differential effects of alendronate and losartan therapy on osteopenia and aortic aneurysm in mice with severe Marfan syndrome. Hum Mol Genet 2010; 19: 4790-4798.
- 11. Davis MR, Summers KM. Structure and function of the mammalian fibrillin gene family: implications for human connective tissue diseases. Mol Genet Metab 2012; Aug 3. [Epub ahead of print.]
- 12. Yetman AT, Bornemeier RA, McCrindle BW. Long-term outcome in patients with Marfan syndrome: is aortic dissection the only cause of sudden death? J Am Coll Cardiol 2003; 41: 329-332.
- 13. Bowne SJ, Sullivan LS, Koboldt DC, et al. Identification of disease-causing mutations in autosomal dominant retinitis pigmentosa (adRP) using next-generation DNA sequencing. Invest Ophthalmol Vis Sci 2011; 52: 494-503.
- 14. Hedges DJ, Guettouche T, Yang S, et al. Comparison of three targeted enrichment strategies on the SOLiD sequencing platform. PLoS One 2011; 6: e18595.
- 15. Ku CS, Naidoo N, Pawitan Y. Revisiting Mendelian disorders through exome sequencing. Hum Genet 2011; 129: 351-370.
- 16. De Leeneer K, Hellemans J, De Schrijver J, et al. Massive parallel amplicon sequencing of the breast cancer genes BRCA1 and BRCA2: opportunities, challenges, and limitations. Hum Mutat 2011; 32: 335-344.
- 17. Faivre L, Masurel-Paulet A, Collod-Béroud G, et al. Clinical and molecular study of 320 children with Marfan syndrome and related type I fibrillinopathies in a series of 1009 probands with pathogenic FBN1 mutations. Pediatrics 2009; 123: 391-398.
- 18. Baetens M, Van Laer L, De Leeneer K, et al. Applying massive parallel sequencing to molecular diagnosis of Marfan and Loeys-Dietz syndromes. Hum Mutat 2011; 32: 1053-1062.
- 19. Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet 2011; 12: 87-98.
- 20. Tang N, Suthers G. FBN1. In: Tang N, Suthers G, editors. RCPA [Royal College of Pathologists of Australasia] Catalogue of Genetic Tests and Laboratories. April 2009. http://genetictesting.rcpa.edu.au/component/gene/genetest/FBN1/ (accessed May 2012).
- 21. NCBI, GeneTests. Marfan syndrome. [Online database] http://www.ncbi.nlm.nih.gov/sites/GeneTests/lab/clinical_disease_id/2104?db=genetests (accessed May 2012).
- 22. Summers KM, Xu D, West JA, et al. An integrated approach to management of Marfan syndrome caused by an FBN1 exon 18 mutation in an Australian Aboriginal family. Clin Genet 2004; 65: 66-69.
Abstract
Marfan syndrome is a multisystem disorder of connective tissue that is inherited in an autosomal dominant fashion, and results from mutation of the FBN1 gene on human chromosome 15.
There are a number of conditions of the connective tissue with a similar phenotype that can be confused with Marfan syndrome. Modifications of the diagnostic criteria have recently been published, facilitating the differentiation of Marfan syndrome from these conditions.
It is still difficult to use modern genetic testing for diagnosis because Marfan syndrome can be caused by many different mutations in FBN1, a large gene with 65 coding segments, while mutations in other genes can cause overlapping phenotypes.
Several clinical trials of drug therapy, including the antihypertensive drug losartan, are in progress.